alimta (Pemetrexed disodium) injection, powder, lyophilized, for solution
[Eli Lilly and Company]
DESCRIPTION
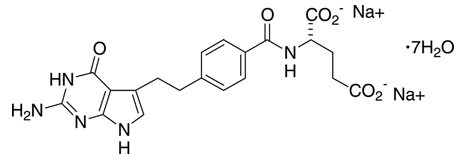
ALIMTA®, pemetrexed for injection, is an antifolate antineoplastic agent that exerts its action by disrupting folate–dependent metabolic processes essential for cell replication. Pemetrexed disodium heptahydrate has the chemical name L-Glutamic acid, N-[4-[2-(2-amino-4,7-dihydro-4-oxo-1H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-, disodium salt, heptahydrate. It is a white to almost–white solid with a molecular formula of C20H19N5Na2O6•7H2O and a molecular weight of 597.49. The structural formula is as follows:
ALIMTA is supplied as a sterile lyophilized powder for intravenous infusion available in single–dose vials. The product is a white to either light yellow or green–yellow lyophilized solid. Each 100 mg or 500 mg vial of ALIMTA contains pemetrexed disodium equivalent to 100 mg pemetrexed and 106 mg mannitol or 500 mg pemetrexed and 500 mg mannitol, respectively. Hydrochloric acid and/or sodium hydroxide may have been added to adjust pH.
CLINICAL PHARMACOLOGY
Pharmacodynamics
Pemetrexed is an antifolate containing the pyrrolopyrimidine–based nucleus that exerts its antineoplastic activity by disrupting folate–dependent metabolic processes essential for cell replication. In vitro studies have shown that pemetrexed inhibits thymidylate synthase (TS), dihydrofolate reductase (DHFR), and glycinamide ribonucleotide formyltransferase (GARFT), all folate–dependent enzymes involved in the de novo biosynthesis of thymidine and purine nucleotides. Pemetrexed is transported into cells by both the reduced folate carrier and membrane folate binding protein transport systems. Once in the cell, pemetrexed is converted to polyglutamate forms by the enzyme folylpolyglutamate synthetase. The polyglutamate forms are retained in cells and are inhibitors of TS and GARFT. Polyglutamation is a time– and concentration–dependent process that occurs in tumor cells and, to a lesser extent, in normal tissues. Polyglutamated metabolites have an increased intracellular half–life resulting in prolonged drug action in malignant cells.
Preclinical studies have shown that pemetrexed inhibits the in vitro growth of mesothelioma cell lines (MSTO–211H, NCI–H2052). Studies with the MSTO–211H mesothelioma cell line showed synergistic effects when pemetrexed was combined concurrently with cisplatin.
Absolute neutrophil counts (ANC) following single–agent administration of pemetrexed to patients not receiving folic acid and vitamin B12 supplementation were characterized using population pharmacodynamic analyses. Severity of hematologic toxicity, as measured by the depth of the ANC nadir, correlates with the systemic exposure of pemetrexed. It was also observed that lower ANC nadirs occurred in patients with elevated baseline cystathionine or homocysteine concentrations. The levels of these substances can be reduced by folic acid and vitamin B12 supplementation. There is no cumulative effect of pemetrexed exposure on ANC nadir over multiple treatment cycles.
Time to ANC nadir with pemetrexed systemic exposure (AUC), varied between 8 to 9.6 days over a range of exposures from 38.3 to 316.8 μg•hr/mL. Return to baseline ANC occurred 4.2 to 7.5 days after the nadir over the same range of exposures.
Pharmacokinetics
The pharmacokinetics of pemetrexed administered as a single agent in doses ranging from 0.2 to 838 mg/m2 infused over a 10–minute period have been evaluated in 426 cancer patients with a variety of solid tumors. Pemetrexed is not metabolized to an appreciable extent and is primarily eliminated in the urine, with 70% to 90% of the dose recovered unchanged within the first 24 hours following administration. The total systemic clearance of pemetrexed is 91.8 mL/min and the elimination half–life of pemetrexed is 3.5 hours in patients with normal renal function (creatinine clearance of 90 mL/min). The clearance decreases, and exposure (AUC) increases, as renal function decreases. Pemetrexed total systemic exposure (AUC) and maximum plasma concentration (Cmax) increase proportionally with dose. The pharmacokinetics of pemetrexed do not change over multiple treatment cycles. Pemetrexed has a steady–state volume of distribution of 16.1 liters. In vitro studies indicate that pemetrexed is approximately 81% bound to plasma proteins. Binding is not affected by degree of renal impairment.
Drug Interactions
Chemotherapeutic Agents — Cisplatin does not affect the pharmacokinetics of pemetrexed and the pharmacokinetics of total platinum are unaltered by pemetrexed.
Vitamins — Coadministration of oral folic acid or intramuscular vitamin B12 does not affect the pharmacokinetics of pemetrexed.
Drugs Metabolized by Cytochrome P450 Enzymes — Results from in vitro studies with human liver microsomes predict that pemetrexed would not cause clinically significant inhibition of metabolic clearance of drugs metabolized by CYP3A, CYP2D6, CYP2C9, and CYP1A2. No studies were conducted to determine the cytochrome P450 isozyme induction potential of pemetrexed, because ALIMTA used as recommended (once every 21 days) would not be expected to cause any significant enzyme induction.
Aspirin — Aspirin, administered in low to moderate doses (325 mg every 6 hours), does not affect the pharmacokinetics of pemetrexed. The effect of greater doses of aspirin on pemetrexed pharmacokinetics is unknown.
Ibuprofen — Daily ibuprofen doses of 400 mg qid reduce pemetrexed’s clearance by about 20% (and increase AUC by 20%) in patients with normal renal function. The effect of greater doses of ibuprofen on pemetrexed pharmacokinetics is unknown (see Drug Interactions under PRECAUTIONS).
Special Populations
The pharmacokinetics of pemetrexed in special populations were examined in about 400 patients in controlled and single arm studies.
Geriatric
No effect of age on the pharmacokinetics of pemetrexed was observed over a range of 26 to 80 years.
Pediatric
Pediatric patients were not included in clinical trials.
Gender
The pharmacokinetics of pemetrexed were not different in male and female patients.
Race
The pharmacokinetics of pemetrexed were similar in Caucasians and patients of African descent. Insufficient data are available to compare pharmacokinetics for other ethnic groups.
Hepatic Insufficiency
There was no effect of elevated AST (SGOT), ALT (SGPT), or total bilirubin on the pharmacokinetics of pemetrexed. However, studies of hepatically impaired patients have not been conducted (see PRECAUTIONS).
Renal Insufficiency
Pharmacokinetic analyses of pemetrexed included 127 patients with reduced renal function. Plasma clearance of pemetrexed decreases as renal function decreases, with a resultant increase in systemic exposure. Patients with creatinine clearances of 45, 50, and 80 mL/min had 65%, 54%, and 13% increases, respectively in pemetrexed total systemic exposure (AUC) compared to patients with creatinine clearance of 100 mL/min (see WARNINGS and DOSAGE AND ADMINISTRATION).
CLINICAL STUDIES
Malignant Pleural Mesothelioma
The safety and efficacy of ALIMTA have been evaluated in chemonaive patients with malignant pleural mesothelioma (MPM) in combination with cisplatin.
Randomized Trial: A multi–center, randomized, single–blind study in 448 chemonaive patients with MPM compared survival in patients treated with ALIMTA in combination with cisplatin to survival in patients receiving cisplatin alone. ALIMTA was administered intravenously over 10 minutes at a dose of 500 mg/m2 and cisplatin was administered intravenously over 2 hours at a dose of 75 mg/m2 beginning approximately 30 minutes after the end of administration of ALIMTA. Both drugs were given on Day 1 of each 21–day cycle. After 117 patients were treated, white cell and GI toxicity led to a change in protocol whereby all patients were given folic acid and vitamin B12 supplementation.
The primary analysis of this study was performed on the population of all patients randomly assigned to treatment who received study drug (randomized and treated). An analysis was also performed on patients who received folic acid and vitamin B12 supplementation during the entire course of study therapy (fully supplemented), as supplementation is recommended (see DOSAGE AND ADMINISTRATION). Results in all patients and those fully supplemented were similar. Patient demographics are shown in Table 1.
|
Patient characteristic |
Randomized and Treated Patients |
Fully Supplemented Patients |
||
|
ALIMTA/cis |
Cisplatin |
ALIMTA/cis |
Cisplatin |
|
|
Age (yrs) | ||||
|
Median (range) |
61 (29–85) |
60 (19–84) |
60 (29–85) |
60 (19–82) |
|
Gender (%) | ||||
|
Male |
184 (81.4) |
181 (81.5) |
136 (81.0) |
134 (82.2) |
|
Female |
42 (18.6) |
41 (18.5) |
32 (19.0) |
29 (17.8) |
|
Origin (%) | ||||
|
Caucasian |
204 (90.3) |
206 (92.8) |
150 (89.3) |
153 (93.9) |
|
Hispanic |
11 (4.9) |
12 (5.4) |
10 (6.0) |
7 (4.3) |
|
Asian |
10 (4.4) |
4 (1.9) |
7 (4.2) |
3 (1.8) |
|
African descent |
1 (0.4) |
0 |
1 (0.6) |
0 |
|
Stage at Entry (%) | ||||
|
I |
16 (7.1) |
14 (6.3) |
15 (8.9) |
12 (7.4) |
|
II |
35 (15.6) |
33 (15.0) |
27 (16.2) |
27 (16.8) |
|
III |
73 (32.4) |
68 (30.6) |
51 (30.5) |
49 (30.4) |
|
IV |
101 (44.9) |
105 (47.2) |
74 (44.3) |
73 (45.3) |
|
Unspecified |
1 (0.4) |
2 (0.9) |
1 (0.6) |
2 (1.2) |
|
Diagnosis/Histology* (%) | ||||
|
Epithelial |
154 (68.1) |
152 (68.5) |
117 (69.6) |
113 (69.3) |
|
Mixed |
37 (16.4) |
36 (16.2) |
25 (14.9) |
25 (15.3) |
|
Sarcomatoid |
18 (8.0) |
25 (11.3) |
14 (8.3) |
17 (10.4) |
|
Other |
17 (7.5) |
9 (4.1) |
12 (7.1) |
8 (4.9) |
|
Baseline KPS† (%) | ||||
|
70–80 |
109 (48.2) |
97 (43.7) |
83 (49.4) |
69 (42.3) |
|
90–100 |
117 (51.8) |
125 (56.3) |
85 (50.6) |
94 (57.7) |
Table 2 summarizes the survival results for all randomized and treated patients regardless of vitamin supplementation status and those patients receiving vitamin supplementation from the time of enrollment in the trial.
|
||||
|
Efficacy Parameter |
Randomized and Treated Patients |
Fully Supplemented Patients |
||
|
ALIMTA/cis |
Cisplatin |
ALIMTA/cis |
Cisplatin |
|
|
Median overall survival |
12.1 mos |
9.3 mos |
13.3 mos |
10.0 mos |
|
Hazard ratio |
0.77 |
0.75 |
||
|
Log rank p–value* |
0.020 |
0.051 |
||
Similar results were seen in the analysis of patients (N=303) with confirmed histologic diagnosis of malignant pleural mesothelioma. Exploratory demographic analyses showed no apparent differences in patients over or under 65. There were too few non–white patients to assess possible ethnic differences. The effect in women (median survival 15.7 months with the combination vs. 7.5 months on cisplatin alone), however, was larger than the effect in males (median survival 11 vs. 9.4 respectively). As with any exploratory analysis, it is not clear whether this difference is real or is a chance finding.
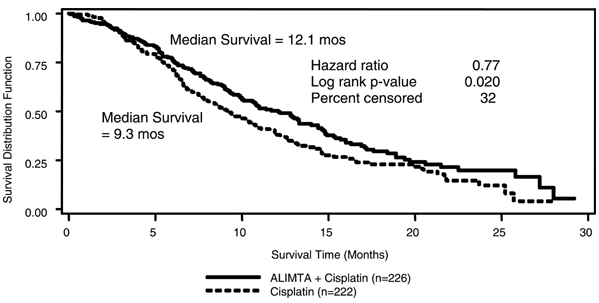
Figure 1: Kaplan-Meier Estimates of Survival Time for ALIMTA plus Cisplatin and Cisplatin Alone in all Randomized and Treated Patients.
Objective tumor response for malignant pleural mesothelioma is difficult to measure and response criteria are not universally agreed upon. However, based upon prospectively defined criteria, the objective tumor response rate for ALIMTA plus cisplatin was greater than the objective tumor response rate for cisplatin alone. There was also improvement in lung function (forced vital capacity) in the ALIMTA plus cisplatin arm compared to the control arm.
Patients who received full supplementation with folic acid and vitamin B12 during study therapy received a median of 6 and 4 cycles in the ALIMTA/cisplatin (N=168) and cisplatin (N=163) arms, respectively. Patients who never received folic acid and vitamin B12 during study therapy received a median of 2 cycles in both treatment arms (N=32 and N=38 for the ALIMTA/cisplatin and cisplatin arm, respectively). Patients receiving ALIMTA in the fully supplemented group received a relative dose intensity of 93% of the protocol specified ALIMTA dose intensity; patients treated with cisplatin in the same group received 94% of the projected dose intensity. Patients treated with cisplatin alone had a dose intensity of 96%.
Non–Small Cell Lung Cancer (NSCLC)
The safety and efficacy of ALIMTA as a single–agent have been evaluated in patients with locally advanced or metastatic (Stage III or IV) non–small cell lung cancer after prior chemotherapy.
Randomized Trial: A multi–center, randomized, open label Phase 3 study was conducted to compare the overall survival following treatment with ALIMTA versus docetaxel. ALIMTA was administered intravenously over 10 minutes at a dose of 500 mg/m2 and docetaxel was administered at 75 mg/m2 as a 1–hour intravenous infusion. Both drugs were given on Day 1 of each 21–day cycle. All patients treated with ALIMTA received vitamin supplementation with folic acid and vitamin B12. The study was intended to show either an overall survival superiority or non–inferiority of ALIMTA to docetaxel. Patient demographics of the intent to treat (ITT) population are shown in Table 3.
|
ALIMTA |
Docetaxel |
|
|
Patient characteristic |
(N=283) |
(N=288) |
|
Age (yrs) | ||
|
Median (range) |
59 (22–81) |
57 (28–87) |
|
Gender (%) | ||
|
Male/Female |
68.6/31.4 |
75.3/24.7 |
|
Stage at Entry (%) | ||
|
III/IV |
25.1/74.9 |
25.3/74.7 |
|
Diagnosis/Histology (%) | ||
|
Adenocarcinoma |
154 (54.4) |
142 (49.3) |
|
Squamous |
78 (27.6) |
93 (32.3) |
|
Bronchoalveolar |
4 (1.4) |
1 (0.3) |
|
Other |
51 (18.1) |
53 (18.5) |
|
Performance Status (%) | ||
|
0–1 |
234 (88.6) |
240 (87.6) |
|
2 |
30 (11.4) |
34 (12.4) |
The primary endpoint in this study was overall survival. The median survival time was 8.3 months in the ALIMTA treatment arm and 7.9 months in the docetaxel arm, with a hazard ratio of 0.99 (see Table 4). The study did not show an overall survival superiority of ALIMTA. Non–inferiority of ALIMTA to docetaxel could not be demonstrated, because a reliable and consistent survival effect of docetaxel required for a non–inferiority analysis could not be estimated from historical trials. In addition, significant treatment crossover at the time of disease progression may have confounded the survival interpretation. The demonstrated surrogate endpoint, response rate allowed the conclusion that an effect of ALIMTA on survival is reasonably likely.
Exploratory demographic analyses on survival showed no significant differences between ALIMTA and docetaxel in patients over or under 65 years of age. There were too few non–white patients to assess possible ethnic differences. Regarding gender, females lived longer than males in both treatment groups. There was no difference in survival between ALIMTA and docetaxel with respect to gender after adjusting for prognostic factors.
Secondary endpoints evaluated in the trial include objective response rate, progression free survival (PFS) and time to progressive disease (TTPD). There was no statistically significant difference between ALIMTA and docetaxel with respect to objective response rate, progression free survival (PFS) and time to progressive disease (TTPD).
|
ALIMTA |
Docetaxel |
|
|
Median overall survival (95% CI) |
8.3 mos (7.0–9.4) |
7.9 mos (6.3–9.2) |
|
Hazard ratio (HR) (95% CI) |
0.99* (0.82–1.20) |
|
|
Log rank p–value |
0.93 |
|
|
1-year survival (95% CI) |
29.7% (23.7–35.6) |
29.7% (23.9–35.5) |
|
Median progression free survival |
2.9 mos |
2.9 mos |
|
Hazard ratio (HR) (95% CI) |
0.97* (0.82–1.16) |
|
|
Time to Progressive Disease |
3.4 mos |
3.5 mos |
|
Hazard ratio (HR) (95% CI) |
0.97* (0.80–1.17) |
|
|
9.1% (5.9–13.2) |
8.8% (5.7–12.8) |
|
INDICATIONS AND USAGE
Mesothelioma
ALIMTA in combination with cisplatin is indicated for the treatment of patients with malignant pleural mesothelioma whose disease is unresectable or who are otherwise not candidates for curative surgery.
Non–Small Cell Lung Cancer
ALIMTA as a single–agent is indicated for the treatment of patients with locally advanced or metastatic non–small cell lung cancer after prior chemotherapy.
The effectiveness of ALIMTA in second–line NSCLC was based on the surrogate endpoint, response rate. There are no controlled trials demonstrating a clinical benefit, such as a favorable survival effect or improvement of disease–related symptoms.
CONTRAINDICATIONS
ALIMTA is contraindicated in patients who have a history of severe hypersensitivity reaction to pemetrexed or to any other ingredient used in the formulation.
WARNINGS
Decreased Renal Function
ALIMTA is primarily eliminated unchanged by renal excretion. No dosage adjustment is needed in patients with creatinine clearance ≥45 mL/min. Insufficient numbers of patients have been studied with creatinine clearance <45 mL/min to give a dose recommendation. Therefore, ALIMTA should not be administered to patients whose creatinine clearance is <45 mL/min (see Dose Reduction Recommendations under DOSAGE AND ADMINISTRATION).
One patient with severe renal impairment (creatinine clearance 19 mL/min) who did not receive folic acid and vitamin B12 died of drug–related toxicity following administration of ALIMTA alone.
Bone Marrow Suppression
ALIMTA can suppress bone marrow function, as manifested by neutropenia, thrombocytopenia, and anemia (or pancytopenia) (see ADVERSE REACTIONS); myelosuppression is usually the dose–limiting toxicity. Dose reductions for subsequent cycles are based on nadir ANC, platelet count, and maximum nonhematologic toxicity seen in the previous cycle (see Dose Reduction Recommendations under DOSAGE AND ADMINISTRATION).
Need for Folate and Vitamin B12 Supplementation
Patients treated with ALIMTA must be instructed to take folic acid and vitamin B12 as a prophylactic measure to reduce treatment–related hematologic and GI toxicity (see DOSAGE AND ADMINISTRATION). In clinical studies, less overall toxicity and reductions in Grade 3/4 hematologic and nonhematologic toxicities such as neutropenia, febrile neutropenia, and infection with Grade 3/4 neutropenia were reported when pretreatment with folic acid and vitamin B12 was administered.
Pregnancy Category D
ALIMTA may cause fetal harm when administered to a pregnant woman. Pemetrexed was fetotoxic and teratogenic in mice at i.p. doses of 0.2 mg/kg (0.6 mg/m2) or 5 mg/kg (15 mg/m2) when given on gestation days 6 through 15. Pemetrexed caused fetal malformations (incomplete ossification of talus and skull bone) at 0.2 mg/kg (about 1/833 the recommended i.v. human dose on a mg/m2 basis), and cleft palate at 5 mg/kg (about 1/33 the recommended i.v. human dose on a mg/m2 basis). Embryotoxicity was characterized by increased embryo–fetal deaths and reduced litter sizes. There are no studies of ALIMTA in pregnant women. Patients should be advised to avoid becoming pregnant. If ALIMTA is used during pregnancy, or if the patient becomes pregnant while taking ALIMTA, the patient should be apprised of the potential hazard to the fetus.
PRECAUTIONS
General
ALIMTA should be administered under the supervision of a qualified physician experienced in the use of antineoplastic agents. Appropriate management of complications is possible only when adequate diagnostic and treatment facilities are readily available. Treatment–related adverse events of ALIMTA seen in clinical trials have been reversible. Skin rash has been reported more frequently in patients not pretreated with a corticosteroid in clinical trials. Pretreatment with dexamethasone (or equivalent) reduces the incidence and severity of cutaneous reaction (see DOSAGE AND ADMINISTRATION).
The effect of third space fluid, such as pleural effusion and ascites, on ALIMTA is unknown. In patients with clinically significant third space fluid, consideration should be given to draining the effusion prior to ALIMTA administration.
Laboratory Tests
Complete blood cell counts, including platelet counts and periodic chemistry tests, should be performed on all patients receiving ALIMTA. Patients should be monitored for nadir and recovery, which were tested in the clinical study before each dose and on days 8 and 15 of each cycle. Patients should not begin a new cycle of treatment unless the ANC is ≥1500 cells/mm3, the platelet count is ≥100,000 cells/mm3, and creatinine clearance is ≥45 mL/min.
Drug Interactions
ALIMTA is primarily eliminated unchanged renally as a result of glomerular filtration and tubular secretion. Concomitant administration of nephrotoxic drugs could result in delayed clearance of ALIMTA. Concomitant administration of substances that are also tubularly secreted (e.g., probenecid) could potentially result in delayed clearance of ALIMTA.
Although ibuprofen (400 mg qid) can be administered with ALIMTA in patients with normal renal function (creatinine clearance ≥80 mL/min), caution should be used when administering ibuprofen concurrently with ALIMTA to patients with mild to moderate renal insufficiency (creatinine clearance from 45 to 79 mL/min). Patients with mild to moderate renal insufficiency should avoid taking NSAIDs with short elimination half–lives for a period of 2 days before, the day of, and 2 days following administration of ALIMTA.
In the absence of data regarding potential interaction between ALIMTA and NSAIDs with longer half–lives, all patients taking these NSAIDs should interrupt dosing for at least 5 days before, the day of, and 2 days following ALIMTA administration. If concomitant administration of an NSAID is necessary, patients should be monitored closely for toxicity, especially myelosuppression, renal, and gastrointestinal toxicity.
Drug/Laboratory Test Interactions
None known.
Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity studies have been conducted with pemetrexed. Pemetrexed was clastogenic in the in vivo micronucleus assay in mouse bone marrow but was not mutagenic in multiple in vitro tests (Ames assay, CHO cell assay). Pemetrexed administered at i.v. doses of 0.1 mg/kg/day or greater to male mice (about 1/1666 the recommended human dose on a mg/m2 basis) resulted in reduced fertility, hypospermia, and testicular atrophy.
Pregnancy
Pregnancy Category D (see WARNINGS).
Nursing Mothers
It is not known whether ALIMTA or its metabolites are excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from ALIMTA, it is recommended that nursing be discontinued if the mother is treated with ALIMTA.
Pediatric Use
The safety and effectiveness of ALIMTA in pediatric patients have not been established.
Geriatric Use
Dose adjustments based on age other than those recommended for all patients have not been necessary (see Special Populations under CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Gender
Dose adjustments based on gender other than those recommended for all patients have not been necessary (see Special Populations under CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Patients with Hepatic Impairment
Patients with bilirubin >1.5 times the upper limit of normal were excluded from clinical trials of ALIMTA. Patients with transaminase >3.0 times the upper limit of normal were routinely excluded from clinical trials if they had no evidence of hepatic metastases. Patients with transaminase from 3 to 5 times the upper limit of normal were included in the clinical trial of ALIMTA if they had hepatic metastases.
Dose adjustments based on hepatic impairment experienced during treatment with ALIMTA are provided in Table 9 (see Special Populations under CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION).
Patients with Renal Impairment
ALIMTA is known to be primarily excreted by the kidney. Decreased renal function will result in reduced clearance and greater exposure (AUC) to ALIMTA compared with patients with normal renal function. Cisplatin coadministration with ALIMTA has not been studied in patients with moderate renal impairment (see Special Populations under CLINICAL PHARMACOLOGY).
ADVERSE REACTIONS
Malignant Pleural Mesothelioma
In Table 5 adverse events occurring in at least 5% of patients are shown along with important effects (renal failure, infection) occurring at lower rates. Adverse events equally or more common in the cisplatin group are not included. The adverse effects more common in the ALIMTA group were primarily hematologic effects, fever and infection, stomatitis/pharyngitis, and rash/desquamation.
|
||||||
|
All Reported Adverse Events Regardless of Causality |
||||||
|
ALIMTA/cis |
Cisplatin |
|||||
|
All Grades |
Grade 3 |
Grade 4 |
All Grades |
Grade 3 |
Grade 4 |
|
|
Laboratory | ||||||
|
Hematologic | ||||||
|
Neutropenia |
58 |
19 |
5 |
16 |
3 |
1 |
|
Leukopenia |
55 |
14 |
2 |
20 |
1 |
0 |
|
Anemia |
33 |
5 |
1 |
14 |
0 |
0 |
|
Thrombocytopenia |
27 |
4 |
1 |
10 |
0 |
0 |
|
Renal | ||||||
|
Creatinine elevation |
16 |
1 |
0 |
12 |
1 |
0 |
|
Renal failure |
2 |
0 |
1 |
1 |
0 |
0 |
|
Clinical | ||||||
|
Constitutional Symptoms | ||||||
|
Fatigue |
80 |
17 |
0 |
74 |
12 |
1 |
|
Fever |
17 |
0 |
0 |
9 |
0 |
0 |
|
Other constitutional symptoms |
11 |
2 |
1 |
8 |
1 |
1 |
|
Cardiovascular General | ||||||
|
Thrombosis/embolism |
7 |
4 |
2 |
4 |
3 |
1 |
|
Gastrointestinal | ||||||
|
Nausea |
84 |
11 |
1 |
79 |
6 |
0 |
|
Vomiting |
58 |
10 |
1 |
52 |
4 |
1 |
|
Constipation |
44 |
2 |
1 |
39 |
1 |
0 |
|
Anorexia |
35 |
2 |
0 |
25 |
1 |
0 |
|
Stomatitis/pharyngitis |
28 |
2 |
1 |
9 |
0 |
0 |
|
Diarrhea without colostomy |
26 |
4 |
0 |
16 |
1 |
0 |
|
Dehydration |
7 |
3 |
1 |
1 |
1 |
0 |
|
Dysphagia/esophagitis/odynophagia |
6 |
1 |
0 |
6 |
0 |
0 |
|
Pulmonary | ||||||
|
Dyspnea |
66 |
10 |
1 |
62 |
5 |
2 |
|
Pain | ||||||
|
Chest pain |
40 |
8 |
1 |
30 |
5 |
1 |
|
Neurology | ||||||
|
Neuropathy/sensory |
17 |
0 |
0 |
15 |
1 |
0 |
|
Mood alteration/depression |
14 |
1 |
0 |
9 |
1 |
0 |
|
Infection/Febrile Neutropenia | ||||||
|
Infection without neutropenia |
11 |
1 |
1 |
4 |
0 |
0 |
|
Infection
with Grade 3 or |
6 |
1 |
0 |
4 |
0 |
0 |
|
Infection/febrile neutropenia–other |
3 |
1 |
0 |
2 |
0 |
0 |
|
Febrile neutropenia |
1 |
1 |
0 |
1 |
0 |
0 |
|
Immune | ||||||
|
Allergic reaction/hypersensitivity |
2 |
0 |
0 |
1 |
0 |
0 |
|
Dermatology/Skin | ||||||
|
Rash/desquamation |
22 |
1 |
0 |
9 |
0 |
0 |
Table 6 compares the incidence (percentage of patients) of CTC Grade 3/4 toxicities in patients who received vitamin supplementation with daily folic acid and vitamin B12 from the time of enrollment in the study (fully supplemented) with the incidence in patients who never received vitamin supplementation (never supplemented) during the study in the ALIMTA plus cisplatin arm.
|
||
|
Adverse Event Regardless of |
Fully Supplemented Patients (N=168) |
Never Supplemented Patients (N=32) |
|
Neutropenia |
24 |
38 |
|
Thrombocytopenia |
5 |
9 |
|
Nausea |
12 |
31 |
|
Vomiting |
11 |
34 |
|
Anorexia |
2 |
9 |
|
Diarrhea without colostomy |
4 |
9 |
|
Dehydration |
4 |
9 |
|
Fever |
0 |
6 |
|
Febrile neutropenia |
1 |
9 |
|
Infection with Grade 3/4 neutropenia |
1 |
6 |
|
Fatigue |
17 |
25 |
The following adverse events were greater in the fully supplemented group compared to the never supplemented group: hypertension (11%, 3%), chest pain (8%, 6%), and thrombosis/embolism (6%, 3%).
For fully supplemented patients treated with ALIMTA plus cisplatin, the incidence of CTC Grade 3/4 fatigue, leukopenia, neutropenia, and thrombocytopenia were greater in patients 65 years or older as compared to patients younger than 65. No relevant effect for ALIMTA safety due to gender or race was identified, except an increased incidence of rash in men (24%) compared to women (16%).
Non–Small Cell Lung Cancer (NSCLC)
Table 7 provides the clinically relevant undesirable effects that have been reported in 265 patients randomly assigned to receive single–agent ALIMTA with folic acid and vitamin B12 supplementation and 276 patients randomly assigned to receive single–agent docetaxel. All patients were diagnosed with locally advanced or metastatic NSCLC and had received prior chemotherapy.
|
||||||
|
All Reported
Adverse Events |
||||||
|
ALIMTA |
Docetaxel |
|||||
|
All Grades |
Grade 3 |
Grade 4 |
All Grades |
Grade 3 |
Grade 4 |
|
|
Laboratory | ||||||
|
Hematologic | ||||||
|
Anemia |
33 |
6 |
2 |
33 |
6 |
<1 |
|
Leukopenia |
13 |
4 |
<1 |
34 |
17 |
11 |
|
Neutropenia |
11 |
3 |
2 |
45 |
8 |
32 |
|
Thrombocytopenia |
9 |
2 |
0 |
1 |
1 |
0 |
|
Hepatic/Renal | ||||||
|
ALT elevation |
10 |
2 |
1 |
2 |
<1 |
0 |
|
AST elevation |
8 |
<1 |
1 |
1 |
<1 |
0 |
|
Decreased creatinine clearance |
5 |
1 |
0 |
1 |
0 |
0 |
|
Creatinine elevation |
3 |
0 |
0 |
1 |
0 |
0 |
|
Renal failure |
<1 |
0 |
0 |
<1 |
0 |
0 |
|
Clinical | ||||||
|
Constitutional Symptoms | ||||||
|
Fatigue |
87 |
14 |
2 |
81 |
16 |
1 |
|
Fever |
26 |
1 |
<1 |
19 |
<1 |
0 |
|
Edema |
19 |
<1 |
0 |
24 |
<1 |
0 |
|
Myalgia |
13 |
2 |
0 |
20 |
3 |
0 |
|
Alopecia |
11 |
NA |
NA |
42 |
NA |
NA |
|
Arthralgia |
8 |
<1 |
0 |
13 |
3 |
0 |
|
Other constitutional symptoms |
8 |
1 |
1 |
6 |
1 |
<1 |
|
Cardiovascular General | ||||||
|
Thrombosis/embolism |
4 |
2 |
1 |
3 |
2 |
1 |
|
Cardiac ischemia |
3 |
2 |
1 |
2 |
<1 |
0 |
|
Gastrointestinal | ||||||
|
Anorexia |
62 |
4 |
1 |
58 |
7 |
<1 |
|
Nausea |
39 |
4 |
0 |
25 |
3 |
0 |
|
Constipation |
30 |
0 |
0 |
23 |
1 |
0 |
|
Vomiting |
25 |
2 |
0 |
19 |
1 |
0 |
|
Diarrhea without colostomy |
21 |
<1 |
0 |
34 |
4 |
0 |
|
Stomatitis/pharyngitis |
20 |
1 |
0 |
23 |
1 |
0 |
|
Dysphagia/esophagitis/odynophagia |
5 |
1 |
<1 |
7 |
1 |
0 |
|
Dehydration |
3 |
1 |
0 |
4 |
1 |
0 |
|
Pulmonary | ||||||
|
Dyspnea |
72 |
14 |
4 |
74 |
17 |
9 |
|
Pain | ||||||
|
Chest pain |
38 |
6 |
<1 |
32 |
7 |
<1 |
|
Neurology | ||||||
|
Neuropathy/sensory |
29 |
2 |
0 |
32 |
1 |
0 |
|
Mood alteration/depression |
11 |
0 |
<1 |
10 |
1 |
0 |
|
Infection/Febrile Neutropenia | ||||||
|
Infection without neutropenia |
23 |
5 |
<1 |
17 |
3 |
1 |
|
Infection/febrile neutropenia–other |
6 |
2 |
0 |
2 |
<1 |
0 |
|
Febrile neutropenia |
2 |
1 |
1 |
14 |
10 |
3 |
|
Infection
with Grade 3 or |
<1 |
0 |
0 |
6 |
4 |
1 |
|
Immune | ||||||
|
Allergic reaction/hypersensitivity |
8 |
0 |
0 |
8 |
1 |
<1 |
|
Dermatology/Skin | ||||||
|
Rash/desquamation |
17 |
0 |
0 |
9 |
0 |
0 |
Clinically relevant Grade 3 and Grade 4 laboratory toxicities were similar between integrated Phase 2 results from three single–agent ALIMTA studies (N=164) and the Phase 3 single–agent ALIMTA study described above, with the exception of neutropenia (12.8% versus 5.3%, respectively) and alanine transaminase elevation (15.2% versus 1.9%, respectively). These differences were likely due to differences in the patient population, since the Phase 2 studies included chemonaive and heavily pretreated breast cancer patients with pre–existing liver metastases and/or abnormal baseline liver function tests.
The incidence of CTC Grade 3/4 hypertension was the only finding demonstrating an age difference in patients treated with ALIMTA and was greater in patients 65 years or older as compared to younger patients. There are insufficient numbers of non–white patients to assess ethnic differences. The incidence of CTC Grade 3/4 dyspnea was higher in males for both treatment arms.
Post–marketing experience
The following adverse events have been identified during post–approval use of ALIMTA. These events have occurred with ALIMTA when used as a single–agent and in combination therapies. Decisions to include these events are based on the seriousness of the event, frequency of reporting, or potential causal connection to ALIMTA.
Gastrointestinal — Rare cases of colitis have been reported in patients treated with ALIMTA.
Injury, poisoning, and procedural complications — Rare cases of radiation recall have been reported in patients who have previously received radiotherapy.
Respiratory — Rare cases of interstitial pneumonitis have been reported in patients treated with ALIMTA.
OVERDOSAGE
There have been few cases of ALIMTA overdose. Reported toxicities included neutropenia, anemia, thrombocytopenia, mucositis, and rash. Anticipated complications of overdose include bone marrow suppression as manifested by neutropenia, thrombocytopenia, and anemia. In addition, infection with or without fever, diarrhea, and mucositis may be seen. If an overdose occurs, general supportive measures should be instituted as deemed necessary by the treating physician.
In clinical trials, leucovorin was permitted for CTC Grade 4 leukopenia lasting ≥3 days, CTC Grade 4 neutropenia lasting ≥3 days, and immediately for CTC Grade 4 thrombocytopenia, bleeding associated with Grade 3 thrombocytopenia, or Grade 3 or 4 mucositis. The following intravenous doses and schedules of leucovorin were recommended for intravenous use: 100 mg/m2, intravenously once, followed by leucovorin, 50 mg/m2, intravenously every 6 hours for 8 days.
The ability of ALIMTA to be dialyzed is unknown.
DOSAGE AND ADMINISTRATION
ALIMTA is for Intravenous Infusion Only
Combination Use With Cisplatin
Malignant Pleural Mesothelioma — The recommended dose of ALIMTA is 500 mg/m2 administered as an intravenous infusion over 10 minutes on Day 1 of each 21–day cycle. The recommended dose of cisplatin is 75 mg/m2 infused over 2 hours beginning approximately 30 minutes after the end of ALIMTA administration. Patients should receive appropriate hydration prior to and/or after receiving cisplatin. See cisplatin package insert for more information.
Single–Agent Use
Non–Small Cell Lung Cancer — The recommended dose of ALIMTA is 500 mg/m2 administered as an intravenous infusion over 10 minutes on Day 1 of each 21–day cycle.
Premedication Regimen
Corticosteroid — Skin rash has been reported more frequently in patients not pretreated with a corticosteroid. Pretreatment with dexamethasone (or equivalent) reduces the incidence and severity of cutaneous reaction. In clinical trials, dexamethasone 4 mg was given by mouth twice daily the day before, the day of, and the day after ALIMTA administration.
Vitamin Supplementation — To reduce toxicity, patients treated with ALIMTA must be instructed to take a low–dose oral folic acid preparation or multivitamin with folic acid on a daily basis. At least 5 daily doses of folic acid must be taken during the 7–day period preceding the first dose of ALIMTA; and dosing should continue during the full course of therapy and for 21 days after the last dose of ALIMTA. Patients must also receive one (1) intramuscular injection of vitamin B12 during the week preceding the first dose of ALIMTA and every 3 cycles thereafter. Subsequent vitamin B12 injections may be given the same day as ALIMTA. In clinical trials, the dose of folic acid studied ranged from 350 to 1000 μg, and the dose of vitamin B12 was 1000 μg. The most commonly used dose of oral folic acid in clinical trials was 400 μg (see WARNINGS).
Laboratory Monitoring and Dose Reduction Recommendations
Monitoring — Complete blood cell counts, including platelet counts, should be performed on all patients receiving ALIMTA. Patients should be monitored for nadir and recovery, which were tested in the clinical study before each dose and on days 8 and 15 of each cycle. Patients should not begin a new cycle of treatment unless the ANC is ≥1500 cells/mm3, the platelet count is ≥100,000 cells/mm3, and creatinine clearance is ≥45 mL/min. Periodic chemistry tests should be performed to evaluate renal and hepatic function.
Dose Reduction Recommendations — Dose adjustments at the start of a subsequent cycle should be based on nadir hematologic counts or maximum nonhematologic toxicity from the preceding cycle of therapy. Treatment may be delayed to allow sufficient time for recovery. Upon recovery, patients should be retreated using the guidelines in Tables 8–10, which are suitable for using ALIMTA as a single agent or in combination with cisplatin.
|
Nadir ANC <500/mm3 and nadir platelets ≥50,000/mm3. |
75% of previous dose (both drugs). |
|
Nadir platelets <50,000/mm3 regardless of nadir ANC. |
50% of previous dose (both drugs). |
If patients develop nonhematologic toxicities (excluding neurotoxicity)≥Grade 3 (except Grade 3 transaminase elevations), ALIMTA should be withheld until resolution to less than or equal to the patient’s pre–therapy value. Treatment should be resumed according to guidelines in Table 9.
|
Dose of ALIMTA |
Dose of Cisplatin |
|
|
Any Grade 3‡ or 4 toxicities except mucositis |
75% of previous dose |
75% of previous dose |
|
Any diarrhea requiring hospitalization (irrespective of Grade) or Grade 3 or 4 diarrhea |
75% of previous dose |
75% of previous dose |
|
Grade 3 or 4 mucositis |
50% of previous dose |
100% of previous dose |
In the event of neurotoxicity, the recommended dose adjustments for ALIMTA and cisplatin are described in Table 10. Patients should discontinue therapy if Grade 3 or 4 neurotoxicity is experienced.
|
Dose of ALIMTA |
Dose of Cisplatin |
|
|
CTC Grade |
(mg/m2) |
(mg/m2) |
|
0–1 |
100% of previous dose |
100% of previous dose |
|
2 |
100% of previous dose |
50% of previous dose |
ALIMTA therapy should be discontinued if a patient experiences any hematologic or nonhematologic Grade 3 or 4 toxicity after 2 dose reductions (except Grade 3 transaminase elevations) or immediately if Grade 3 or 4 neurotoxicity is observed.
Elderly Patients — No dose reductions other than those recommended for all patients are necessary for patients ≥65 years of age.
Children — ALIMTA is not recommended for use in children, as safety and efficacy have not been established in children.
Renally Impaired Patients — In clinical studies, patients with creatinine clearance ≥45 mL/min required no dose adjustments other than those recommended for all patients. Insufficient numbers of patients with creatinine clearance below 45 mL/min have been treated to make dosage recommendations for this group of patients. Therefore, ALIMTA should not be administered to patients whose creatinine clearance is <45 mL/min using the standard Cockcroft and Gault formula (below) or GFR measured by Tc99m–DPTA serum clearance method:
|
Males: |
[140 – Age in years] × Actual Body Weight (kg) |
= mL/min |
|
72 × Serum Creatinine (mg/dL) |
||
|
Females: |
Estimated creatinine clearance for males × 0.85 |
Caution should be exercised when administering ALIMTA concurrently with NSAIDs to patients whose creatinine clearance is <80 mL/min (see Drug Interactions under PRECAUTIONS).
Hepatically Impaired Patients — ALIMTA is not extensively metabolized by the liver. Dose adjustments based on hepatic impairment experienced during treatment with ALIMTA are provided in Table 9 (see Patients with Hepatic Impairment under PRECAUTIONS).
Preparation and Administration Precautions
As with other potentially toxic anticancer agents, care should be exercised in the handling and preparation of infusion solutions of ALIMTA. The use of gloves is recommended. If a solution of ALIMTA contacts the skin, wash the skin immediately and thoroughly with soap and water. If ALIMTA contacts the mucous membranes, flush thoroughly with water. Several published guidelines for handling and disposal of anticancer agents are available.1–4 ALIMTA is not a vesicant. There is no specific antidote for extravasation of ALIMTA. To date, there have been few reported cases of ALIMTA extravasation, which were not assessed as serious by the investigator. ALIMTA extravasation should be managed with local standard practice for extravasation as with other non–vesicants.
Preparation for Intravenous Infusion Administration
-
Use aseptic technique during the reconstitution and further dilution of ALIMTA for intravenous infusion administration.
-
Calculate the dose of ALIMTA and determine the number of vials needed. Vials contain either 100 mg or 500 mg of ALIMTA. The vials contain an excess of ALIMTA to facilitate delivery of label amount.
-
Reconstitute each 100–mg vial with 4.2 mL of 0.9% Sodium Chloride Injection (preservative free). Reconstitute 500–mg vial with 20 mL of 0.9% Sodium Chloride Injection (preservative free). Reconstitution of either size vial gives a solution containing 25 mg/mL ALIMTA. Gently swirl each vial until the powder is completely dissolved. The resulting solution is clear and ranges in color from colorless to yellow or green–yellow without adversely affecting product quality. The pH of the reconstituted ALIMTA solution is between 6.6 and 7.8. FURTHER DILUTION IS REQUIRED.
-
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. If particulate matter is observed, do not administer.
-
An appropriate quantity of the reconstituted ALIMTA solution must be further diluted into a solution of 0.9% Sodium Chloride Injection (preservative free), so that the total volume of solution is 100 mL. ALIMTA is administered as an intravenous infusion over 10 minutes.
-
Chemical and physical stability of reconstituted and infusion solutions of ALIMTA were demonstrated for up to 24 hours following initial reconstitution, when stored at refrigerated or ambient room temperature [see USP Controlled Room Temperature] and lighting. When prepared as directed, reconstitution and infusion solutions of ALIMTA contain no antimicrobial preservatives. Discard any unused portion.
Reconstitution and further dilution prior to intravenous infusion is only recommended with 0.9% Sodium Chloride Injection (preservative free). ALIMTA is physically incompatible with diluents containing calcium, including Lactated Ringer’s Injection, USP and Ringer’s Injection, USP and therefore these should not be used. Coadministration of ALIMTA with other drugs and diluents has not been studied, and therefore is not recommended.
HOW SUPPLIED
ALIMTA® vials:.
100 mg lyophilized powder in a 10–mL size sterile single-use vial with flip-off cap individually packaged in a carton. NDC 0002–7640–01 (VL7640)
500 mg lyophilized powder in a 50–mL size sterile single use vial with flip off cap individually packaged in a carton. NDC 0002–7623–01 (VL7623)
Storage
ALIMTA, pemetrexed for injection, should be stored at 25°C (77°F); excursions permitted to 15–30°C (59–86°F) [see USP Controlled Room Temperature].
Chemical and physical stability of reconstituted and infusion solutions of ALIMTA were demonstrated for up to 24 hours following initial reconstitution, when stored refrigerated, 2–8°C (36–46°F), or at 25°C (77°F), excursions permitted to 15–30°C (59–86°F) [see USP Controlled Room Temperature]. When prepared as directed, reconstituted and infusion solutions of ALIMTA contain no antimicrobial preservatives. Discard unused portion.
ALIMTA is not light sensitive.
REFERENCES
-
Preventing Occupational Exposures to Antineoplastic and Other Hazardous Drugs in Health Care Settings. NIOSH Alert 2004–165.
-
OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
-
American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006;63:1172-1193.
-
Polovich, M., White, J. M., & Kelleher, L. O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
Literature revised September 12, 2007
Eli Lilly and Company
Indianapolis, IN 46285, USA
www.ALIMTA.com
Copyright © 2004, 2007, Eli Lilly and Company. All rights reserved.
Supplement Patient Material Section
INFORMATION
FOR PATIENTS AND CAREGIVERS
ALIMTA® (uh–LIM–tuh)
(pemetrexed for injection)
Read the Patient Information that comes with ALIMTA before you start treatment and each time you get treated with ALIMTA. There may be new information. This leaflet does not take the place of talking to your doctor about your medical condition or treatment. Talk to your doctor if you have any questions about ALIMTA.
What is ALIMTA?
ALIMTA is a treatment for:
-
Malignant pleural mesothelioma. This cancer affects the inside lining of the chest cavity. ALIMTA is given with cisplatin, another anti–cancer medicine (chemotherapy).
-
Non–small cell lung cancer. This cancer is a disease in which malignant (cancer) cells form in the tissues of the lung.
To lower your chances of side effects of ALIMTA, you must also take folic acid and vitamin B12 prior to and during your treatment with ALIMTA. Your doctor will prescribe a medicine called a “corticosteroid” to take for 3 days during your treatment with ALIMTA. Corticosteroid medicines lower your chances of getting skin reactions with ALIMTA.
ALIMTA has not been studied in children.
What should I tell my doctor before taking ALIMTA?
Tell your doctor about all of your medical conditions, including if you:
-
are pregnant or planning to become pregnant. ALIMTA may harm your unborn baby.
-
are breastfeeding. It is not known if ALIMTA passes into breast milk. You should stop breastfeeding once you start treatment with ALIMTA.
-
are taking other medicines, including prescription and nonprescription medicines, vitamins, and herbal supplements. ALIMTA and other medicines may affect each other causing serious side effects. Especially, tell your doctor if you are taking medicines called “nonsteroidal anti–inflammatory drugs” (NSAIDs) for pain or swelling. There are many NSAID medicines. If you are not sure, ask your doctor or pharmacist if any of your medicines are NSAIDs.
How is ALIMTA given?
-
ALIMTA is slowly infused (injected) into a vein. The injection or infusion will last about 10 minutes. You will usually receive ALIMTA once every 21 days (3 weeks).
-
If you are being treated for malignant pleural mesothelioma, ALIMTA is given in combination with cisplatin (another anti–cancer drug). Cisplatin is infused in your vein for about 2 hours starting about 30 minutes after your treatment with ALIMTA.
-
Your doctor will prescribe a medicine called a“corticosteroid” to take for 3 days during your treatment with ALIMTA. Corticosteroid medicines lower your chances for getting skin reactions with ALIMTA.
-
It is very important to take folic acid and vitamin B12 during your treatment with ALIMTA to lower your chances of harmful side effects. You must start taking 350–1000 micrograms of folic acid every day for at least 5 days out of the 7 days before your first dose of ALIMTA. You must keep taking folic acid every day during the time you are getting treatment with ALIMTA, and for 21 days after your last treatment. You can get folic acid vitamins over–the–counter. Folic acid is also found in many multivitamin pills. Ask your doctor or pharmacist for help if you are not sure how to choose a folic acid product. Your doctor will give you vitamin B12 injections while you are getting treatment with ALIMTA. You will get your first vitamin B12 injection during the week before your first dose of ALIMTA, and then about every 9 weeks during treatment.
-
You will have regular blood tests before and during your treatment with ALIMTA. Your doctor may adjust your dose of ALIMTA or delay treatment based on the results of your blood tests and on your general condition.
What should I avoid while taking ALIMTA?
-
Women who can become pregnant should not become pregnant during treatment with ALIMTA. ALIMTA may harm the unborn baby.
-
Ask your doctor before taking medicines called NSAIDs. There are many NSAID medicines. If you are not sure, ask your doctor or pharmacist if any of your medicines are NSAIDs.
What are the possible side effects of ALIMTA?
Most patients taking ALIMTA will have side effects. Sometimes it is not always possible to tell whether ALIMTA, another medicine, or the cancer itself is causing these side effects. Call your doctor right away if you have a fever, chills, diarrhea, or mouth sores. These symptoms could mean you have an infection.
The most common side effects of ALIMTA when given alone or in combination with cisplatin are:
-
Stomach upset, including nausea, vomiting, and diarrhea. You can obtain medicines to help control some of these symptoms. Call your doctor if you get any of these symptoms.
-
Low blood cell counts:
-
Low red blood cells. Low red blood cells may make you feel tired, get tired easily, appear pale, and become short of breath.
-
Low white blood cells. Low white blood cells may give you a greater chance for infection. If you have a fever (temperature above 100.4°F) or other signs of infection, call your doctor right away.
-
Low platelets. Low platelets give you a greater chance for bleeding. Your doctor will do blood tests to check your blood counts before and during treatment with ALIMTA.
-
-
Tiredness. You may feel tired or weak for a few days after your ALIMTA treatments. If you have severe weakness or tiredness, call your doctor.
-
Mouth, throat, or lip sores (stomatitis, pharyngitis). You may get redness or sores in your mouth, throat, or on your lips. These symptoms may happen a few days after ALIMTA treatment. Talk with your doctor about proper mouth and throat care.
-
Loss of appetite. You may lose your appetite and lose weight during your treatment. Talk to your doctor if this is a problem for you.
-
Rash. You may get a rash or itching during treatment. These usually appear between treatments with ALIMTA and usually go away before the next treatment. Call your doctor if you get a severe rash or itching.
Talk with your doctor, nurse or pharmacist about any side effect that bothers you or that doesn’t go away.
These are not all the side effects of ALIMTA. For more information, ask your doctor, nurse or pharmacist.
General information about ALIMTA
Medicines are sometimes prescribed for conditions other than those listed in patient information leaflets. ALIMTA was prescribed for your medical condition.
This leaflet summarizes the most important information about ALIMTA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about ALIMTA that is written for health professionals. You can also call 1–800–LILLY–RX (1–800–545–5979) or visit www.ALIMTA.com.
Literature revised September 12, 2007
Eli Lilly and Company
Indianapolis, IN 46285, USA
www.ALIMTA.com
Copyright © 2004, 2007, Eli Lilly and Company. All rights reserved.
| Alimta (Pemetrexed disodium) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Alimta (Pemetrexed disodium) | ||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
Revised: 10/2007Eli Lilly and Company