AMEVIVE
-
alefacept
Astellas Pharma US Inc.
----------
|
|||||||||||||||||
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
AMEVIVE® is indicated for the treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy.
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Instructions
The recommended dose of AMEVIVE® is 15 mg intramuscularly once weekly for 12 weeks. The CD4+ T lymphocyte counts should be measured before initiating dosing.
AMEVIVE® therapy should not be initiated in patients who have CD4+ T lymphocyte counts below normal. The CD4+ T lymphocyte counts of patients receiving AMEVIVE® should be monitored every two weeks throughout the course of the 12-week dosing regimen. If CD4+ T lymphocyte counts are below 250 cells/µL, AMEVIVE® dosing should be withheld and weekly monitoring instituted. AMEVIVE® should be discontinued if the counts remain below 250 cells/µL for one month [see Warnings and Precautions (5.1)].
An additional 12-week course may be initiated if at least 12-weeks have passed since the previous treatment course and the CD4+ T lymphocyte counts are normal.
2.2 Preparation Instructions
- AMEVIVE® should be reconstituted using aseptic technique. Each vial is for single patient use only.
- AMEVIVE® 15 mg lyophilized powder should only be reconstituted with the supplied diluent (Sterile Water for Injection
- Do not add other medications to solutions containing AMEVIVE®. Do not filter reconstituted solution during preparation or administration.
- Using the supplied syringe and one of the supplied needles, withdraw 0.6 mL of the diluent. Keeping the needle pointed at the sidewall of the vial, slowly inject the diluent into the vial of AMEVIVE®. Foaming will occur. Gently swirl the contents during dissolution. To avoid excessive foaming, do not shake or vigorously agitate. Dissolution of AMEVIVE® generally takes less than two minutes.
- Inspect AMEVIVE® reconstituted solution visually for particulate matter and discoloration. AMEVIVE® reconstituted solution should be clear and colorless to slightly yellow. The solution should not be used if it is cloudy, if there is pronounced discoloration, or if particulate matter is present.
- The reconstituted product should be used immediately or within 4 hours if stored in the vial at 2-8°C (36-46°F). Discard AMEVIVE® not used within 4 hours of reconstitution.
2.3 Administration Instructions
- The reconstituted solution should be clear and colorless to slightly yellow. Do not administer the solution if cloudy or discolored or if undissolved material or particulate matter is present.
- Remove the needle used for reconstitution and attach the other supplied needle. Withdraw 0.5 mL of the AMEVIVE® solution into the syringe, and inject the full 0.5 mL of solution intramuscularly. A volume of 0.5 mL of the reconstituted solution contains 15 mg of alefacept. Rotate injection sites so that a different site is used for each new injection. New injections should be given at least one inch from a previous injection site and never into tender, bruised, erythematous, or indurated skin.
- Discard empty vials of AMEVIVE®.
3 DOSAGE FORMS AND STRENGTHS
For injection; AMEVIVE® is supplied as 15 mg of lyophilized powder in a single-use vial for reconstitution with Sterile Water for Injection, USP.
4 CONTRAINDICATIONS
AMEVIVE® should not be administered to patients infected with HIV. AMEVIVE® reduces CD4+ T lymphocyte counts, which might accelerate disease progression or increase complications of disease in these patients [see Warnings and Precautions (5.1, 5.3)].
5 WARNINGS AND PRECAUTIONS
5.1 Lymphopenia
AMEVIVE® induces dose-dependent reductions in circulating CD4+ and CD8+ T lymphocyte counts. CD4+ counts should be normal before initiating treatment with AMEVIVE® and should be closely monitored during AMEVIVE® treatment [see Dosage and Administration (2.1)]
5.2 Malignancies
AMEVIVE® may increase the risk of malignancies. Malignancies were reported in subjects who received AMEVIVE® in clinical studies [see Adverse Reactions (6.1, 6.3)].
In preclinical studies, animals developed B cell hyperplasia, and one animal developed a lymphoma [see Nonclinical Toxicology (13.1)]. AMEVIVE® should not be administered to patients with a history of systemic malignancy.
Exercise caution when considering the use of AMEVIVE® in patients at high risk for malignancy. Discontinue AMEVIVE® if a patient develops a malignancy.
5.3 Serious Infections
AMEVIVE® is an immunosuppressive agent and, therefore, has the potential to increase the risk of infection and reactivate latent, chronic infections. Serious infections (infections requiring hospitalization) were reported in subjects who received AMEVIVE® [see Adverse Reactions (6.1, 6.3)]. AMEVIVE® should not be administered to patients with a clinically important infection. Exercise caution when considering the use of AMEVIVE® in patients with chronic infections or a history of recurrent infection. Patients should be monitored for signs and symptoms of infection during or after a course of AMEVIVE®. New infections should be closely monitored. If a patient develops a serious infection, AMEVIVE® should be discontinued [see Adverse Reactions (6.1)].
5.4 Concomitant Therapies
Patients receiving other immunosuppressive agents or phototherapy should not receive concurrent therapy with AMEVIVE® because of the possibility of excessive immunosuppression.
5.5 Immunizations
The safety and efficacy of live or live-attenuated vaccines in patients being treated with AMEVIVE® have not been studied. In a study of 46 patients with chronic plaque psoriasis, the ability to mount immunity to tetanus toxoid (recall antigen) and an experimental neo-antigen was preserved in those patients undergoing AMEVIVE® therapy.
5.6 Hypersensitivity Reactions
Urticaria and angioedema have been associated with the administration of AMEVIVE®. If an anaphylactic reaction or other serious allergic reaction occurs, discontinue AMEVIVE® immediately and initiate appropriate therapy.
5.7 Hepatic Injury
In post-marketing experience there have been reports of liver injury, including asymptomatic transaminase elevation, fatty infiltration of the liver, hepatitis, decompensation of cirrhosis with liver failure, and acute liver failure. Liver failure has been reported with concomitant alcohol use [see Adverse Reactions (6.3)].
In the 24-week period constituting the first course of placebo-controlled studies, 1.7% (15/876) of AMEVIVE®-treated patients and 1.2% (5/413) of the placebo group experienced ALT and/or AST elevations of at least 3 times the upper limit of normal. While the exact relationship of these occurrences with the use of AMEVIVE® has not been established, patients with signs or symptoms of liver injury should be fully evaluated. AMEVIVE® should be discontinued in patients who develop clinical signs of liver injury.
6 ADVERSE REACTIONS
The most serious adverse reactions described elsewhere in the labeling include the following:
- Lymphopenia [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
- Serious Infections requiring hospitalization [see Warnings and Precautions (5.3)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Commonly observed adverse events seen in the first course of placebo-controlled clinical trials with at least a 2% higher incidence in the AMEVIVE® -treated patients compared to placebo-treated patients were: pharyngitis, dizziness, increased cough, nausea, pruritus, myalgia, chills, injection site pain, injection site inflammation, and accidental injury. The only adverse event that occurred at a 5% or higher incidence among AMEVIVE®-treated patients compared to placebo-treated patients was chills (1% placebo vs. 6% AMEVIVE®), which occurred predominantly with intravenous administration.
The adverse reactions which most commonly resulted in clinical intervention were cardiovascular events including coronary artery disorder in <1% of subjects and myocardial infarct in <1% of subjects. These events were not observed in any of the 413 placebo-treated subjects. The total number of subjects hospitalized for cardiovascular events in the AMEVIVE®-treated group was 1.2% (11/876).
The most common events resulting in discontinuation of treatment with AMEVIVE® were CD4+ T lymphocyte levels below 250 cells/µL [see Warnings and Precautions (5.1)], headache (0.2%), and nausea (0.2%).
The data described below reflect exposure to AMEVIVE® in a total of 1869 psoriasis patients, of whom 1315 (70%) received 1 to 2 courses of therapy and 554 (30%) received 3 or more courses. The median duration of follow-up was 8.4 months for the patients who received 1 to 2 courses and 27.7 months for the patients who received 3 or more courses of AMEVIVE®. Of the 1869 total patients, 876 received their first course in placebo-controlled studies. The population studied ranged in age from 16 to 84 years, and included 69% men and 31% women. The patients were mostly Caucasian (88%), reflecting the general psoriatic population. Disease severity at baseline was moderate to severe psoriasis.
Effect on Lymphocyte Counts
In the intramuscular study (Study 2), 4% of patients temporarily discontinued treatment and no patients permanently discontinued treatment due to CD4+ T lymphocyte counts below the specified threshold of 250 cells/µL. In Study 2, 10%, 28%, and 42% of patients had total lymphocyte, CD4+, and CD8+ T lymphocyte counts below normal, respectively. Twelve weeks after a course of therapy (12 weekly doses), 2%, 8%, and 21% of patients had total lymphocyte, CD4+, and CD8+ T cell counts below normal.
In the first course of the intravenous study (Study 1), 10% of patients temporarily discontinued treatment and 2% permanently discontinued treatment due to CD4+ T lymphocyte counts below the specified threshold of 250 cells/µL. During the first course of Study 1, 22% of patients had total lymphocyte counts below normal, 48% had CD4+ T lymphocyte counts below normal and 59% had CD8+ T lymphocyte counts below normal. The maximal effect on lymphocytes was observed within 6 to 8 weeks of initiation of treatment. Twelve weeks after a course of therapy (12 weekly doses), 4% of patients had total lymphocyte counts below normal, 19% had CD4+ T lymphocyte counts below normal, and 36% had CD8+ T lymphocyte counts below normal.
For patients receiving a second course of AMEVIVE® in Study 1, 17% of patients had total lymphocyte counts below normal, 44% had CD4+ T lymphocyte counts below normal, and 56% had CD8+ T lymphocyte counts below normal. Twelve weeks after completing dosing, 3% of patients had total lymphocyte counts below normal, 17% had CD4+ T lymphocyte counts below normal, and 35% had CD8+ T lymphocyte counts below normal [see Warnings and Precautions (5.7)].
In an open-label postmarketing study, subjects with psoriasis were treated with up to three courses of Amevive: each course consisted of Amevive 15 mg intramuscular weekly for twelve weeks, followed by twelve weeks of observation. Lymphocyte counts were assessed at regular intervals during the post-treatment observation period. For subjects whose counts went below 75% of the baseline at any time after the last dose in the study, the time from the last dose to the time that their lymphocyte count returned to ≥ 75% of baseline was analyzed. Of 115 evaluable subjects for total lymphocyte counts, the median time of recovery was 2.1 months with a range of 0.6 to 11.1 months. Of the 123 evaluable subjects for CD4+ T cell counts, the median time to recovery was 2.3 months with the range of 0.6 to 12.4 months. Of the 105 evaluable subjects for CD8+ T cell counts, the median time to recovery was 1.6 months with a range of 0.6 to 8.7 months.
Malignancies
In the 24-week period constituting the first course of placebo-controlled studies, 13 malignancies were diagnosed in 11 AMEVIVE®-treated patients. The incidence of malignancies was 1.3% (11/876) for AMEVIVE®-treated patients compared to 0.5% (2/413) in the placebo group.
Among 1869 patients who received AMEVIVE® at any dose in clinical trials, 43 patients were diagnosed with 63 treatment-emergent malignancies. The majority of the malignancies were non-melanoma skin cancers: 46 cases (20 basal cell, 26 squamous cell carcinomas) in 27 patients. Other malignancies observed in AMEVIVE®-treated patients included melanoma (n=3), solid organ malignancies (n=12 in 11 patients), and lymphomas (n=5); the latter consisted of two Hodgkin’s and two non-Hodgkin’s lymphomas, and one cutaneous T cell lymphoma (mycosis fungoides).
Infections
In the 24-week period constituting the first course of placebo-controlled studies, serious infections (infections requiring hospitalization) were seen at a rate of 0.9% (8/876) in AMEVIVE®-treated patients and 0.2% (1/413) in the placebo group. In patients receiving repeated courses of AMEVIVE® therapy, the rates of serious infections remained similar across courses of therapy. Serious infections among 1869 AMEVIVE®-treated patients included cellulitis, abscesses, wound infections, toxic shock, pneumonia, appendicitis, cholecystitis, gastroenteritis and herpes infections.
Hypersensitivity Reactions
In clinical studies, 4 of 1869 (0.2%) patients were reported to experience angioedema: two of these patients were hospitalized. In the 24-week period constituting the first course of placebo-controlled studies, urticaria was reported in 6 (<1%) AMEVIVE®-treated patients vs. 1 patient in the control group. Urticaria resulted in discontinuation of therapy in one of the AMEVIVE®-treated patients.
Hepatic Injury
In the 24-week period constituting the first course of placebo-controlled studies, 1.7% (15/876) of AMEVIVE®-treated patients and 1.2% (5/413) of the placebo group experienced ALT and/or AST elevations of at least 3 times the upper limit of normal.
Injection Site Reactions
In the intramuscular study (Study 2), 16% of AMEVIVE®-treated patients and 8% of placebo-treated patients reported injection site reactions. In patients receiving repeated courses of AMEVIVE® intramuscular therapy, the incidence of injection site reactions remained similar across courses of therapy. Reactions at the site of injection were generally mild, typically occurred on single occasions, and included either pain (7%), inflammation (4%), bleeding (4%), edema (2%), non-specific reaction (2%), mass (1%), or skin hypersensitivity (<1%). In the clinical trials, a single case of injection site reaction led to the discontinuation of AMEVIVE®.
6.2 Immunogenicity
Approximately 3% (40/1357) of patients receiving AMEVIVE® developed low-titer antibodies to alefacept as determined by an ELISA. When anti-alefacept antibodies were assessed using a dual specificity Biacore assay, 72% (72/100) of patients receiving AMEVIVE® were positive for anti-alefacept antibodies. The long-term immunogenicity of AMEVIVE® is unknown.
Results are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to alefacept with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of AMEVIVE. Because these reactions are reported voluntary from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Malignancies
In post-marketing experience there have been reports of malignancies including skin, solid organ, lymphomas and leukemias [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
Serious Infections
In post-marketing experience there have been reports of infections including sepsis, opportunistic infections (viral, fungal, bacterial), cellulitis, urinary tract infection (UTI), Clostridium difficile colitis and pharyngitis [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Hepatic Injury
In post-marketing experience there have been reports of asymptomatic transaminase elevation, fatty infiltration of the liver, hepatitis, and severe liver failure [see Warnings and Precautions (5.7) and Adverse Reactions (6.1)].
7 DRUG INTERACTIONS
Drug interaction studies have not been conducted with AMEVIVE®.
7.1 Concomitant Therapies
The safety of AMEVIVE® in combination with immunosuppressive agents or phototherapy has not been evaluated [see Warnings and Precautions (5.4)].
7.2 CYP450 Substrates
The formation of CYP450 enzymes may be suppressed by increased levels of cytokines (e.g., TNFα, IL-1, IL-6, IL-10, IFN) during chronic inflammation. Therefore, a molecule that exerts its pharmacological effect by inhibiting the release of cytokines, such as AMEVIVE®, could normalize the formation of CYP450 enzymes. Upon initiation or discontinuation of AMEVIVE® in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category B
There are no adequate and well-controlled studies of AMEVIVE® in pregnant women. AMEVIVE® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Reproductive toxicology studies have been performed in cynomolgus monkeys at doses up to 5 mg/kg/week (about 62 times the human dose based on body weight) and have revealed no evidence of impaired fertility or harm to the fetus due to AMEVIVE®. No abortifacient or teratogenic effects were observed in cynomolgus monkeys following intravenous bolus injections of AMEVIVE® administered weekly during the period of organogenesis to gestation. AMEVIVE® underwent trans-placental passage and produced in utero exposure in the developing monkeys. In utero, serum levels of exposure in these monkeys were 23% of maternal serum levels. No evidence of fetal toxicity including adverse effects on immune system development was observed in any of these animals.
8.3 Nursing Mothers
It is not known whether AMEVIVE® is excreted in human milk. Because many drugs are excreted in human milk, and because there exists the potential for serious adverse reactions in nursing infants from AMEVIVE®, a decision should be made whether to discontinue nursing while taking the drug or to discontinue the use of the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and efficacy of AMEVIVE® in pediatric patients have not been studied. AMEVIVE® is not indicated for pediatric patients.
8.5 Geriatric Use
Of the 1869 patients who received AMEVIVE® in clinical trials, a total of 129 patients were ≥ 65 years of age and 16 patients were ≥ 75 years of age. No differences in safety or efficacy were observed between older and younger patients, but there were not sufficient data to exclude important differences. Because the incidence of infections and certain malignancies is higher in the elderly population, in general, caution should be used in treating the elderly.
10 OVERDOSAGE
The highest dose tested in humans (0.75 mg/kg intravenous) was associated with chills, headache, arthralgia, and sinusitis within one day of dosing. Patients who have been inadvertently administered an excess of the recommended dose should be closely monitored for effects on total lymphocyte count and CD4+ T lymphocyte count.
11 DESCRIPTION
AMEVIVE® (alefacept) is a CD2-directed LFA-3/Fc fusion protein that consists of the extracellular CD2-binding portion of the human leukocyte function antigen-3 (LFA-3) linked to the Fc (hinge, CH2 and CH3 domains) portion of human IgG1. Alefacept is produced by recombinant DNA technology in a Chinese Hamster Ovary (CHO) mammalian cell expression system. The molecular weight of alefacept is 91.4 kilodaltons.
AMEVIVE® is supplied as a sterile, white-to-off-white, preservative-free, lyophilized powder for intramuscular administration. After reconstitution with 0.6 mL of the supplied Sterile Water for Injection, USP, the solution of AMEVIVE® is clear, with a pH of approximately 6.9.
AMEVIVE® for intramuscular injection contains alefacept (15 mg), citric acid monohydrate (0.06 mg), glycine (5 mg), sodium citrate dehydrate (3.6 mg), and sucrose (12.5 mg) per 0.5 mL of reconstituted solution.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
AMEVIVE® interferes with lymphocyte activation by specifically binding to the lymphocyte antigen, CD2, and inhibiting the interaction between CD2 and its ligand, LFA-3. Activation of T lymphocytes involving the interaction between LFA-3 on antigen-presenting cells and CD2 on T lymphocytes plays a role in the pathophysiology of chronic plaque psoriasis.
AMEVIVE® also causes a reduction in subsets of CD2+ T lymphocytes and circulating total CD4+ and CD8+ T lymphocyte counts. In clinical studies of AMEVIVE®, minor changes in the numbers of circulating cells other than T lymphocytes have been observed.
12.2 Pharmacodynamics
At doses tested in clinical trials, AMEVIVE® therapy resulted in a dose-dependent decrease in circulating total lymphocytes. This reduction predominantly affected the memory effector subset of the CD4+ and CD8+ T lymphocyte compartments (CD4+CD45RO+ and CD8+CD45RO+), the predominant phenotype in psoriatic lesions. Circulating naïve T lymphocyte and natural killer cell counts appeared to be only minimally susceptible to AMEVIVE® treatment, while circulating B lymphocyte counts appeared not to be affected by AMEVIVE® [see Adverse Reactions (6.1)].
12.3 Pharmacokinetics
In patients with moderate to severe plaque psoriasis, following a 7.5 mg intravenous administration, the mean volume of distribution of alefacept was 94 mL/kg, the mean clearance was 0.25 mL/h/kg, and the mean elimination half-life was approximately 270 hours. Following an intramuscular injection, bioavailability was 63%.
The pharmacokinetics of alefacept in pediatric patients have not been studied. The effects of renal or hepatic impairment on the pharmacokinetics of alefacept have not been studied.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a chronic toxicity study, cynomolgus monkeys were dosed weekly for 52 weeks with intravenous alefacept at 1 mg/kg/dose or 20 mg/kg/dose. One animal in the high dose group developed a B-cell lymphoma that was detected after 28 weeks of dosing. Additional animals in both dose groups developed B-cell hyperplasia of the spleen and lymph nodes. One-year post-treatment there was no evidence of alefacept-related lymphoma or B-cell hyperplasia in any of the remaining treated monkeys.
All animals in the study were positive for an endemic primate gammaherpes virus also known as lymphocryptovirus (LCV). Latent LCV infection is generally asymptomatic, but can lead to B-cell lymphomas when animals are immune suppressed.
In a separate study, baboons given 3 doses of alefacept at 1 mg/kg every 8 weeks were found to have centroblast proliferation in B-cell dependent areas in the germinal centers of the spleen following a 116-day washout period.
The role of AMEVIVE® in the development of the lymphoid malignancy and the hyperplasia observed in non-human primates and the relevance to humans is unknown. Immunodeficiency-associated lymphocyte disorders (plasmacytic hyperplasia, polymorphic proliferation, and B-cell lymphomas) occur in patients who have congenital or acquired immunodeficiencies including those resulting from immunosuppressive therapy.
No formal carcinogenicity or fertility studies were conducted.
Mutagenicity studies were conducted in vitro and in vivo; no evidence of mutagenicity was observed.
14 CLINICAL STUDIES
AMEVIVE® was evaluated in two randomized, double-blind, placebo-controlled studies in adults with chronic (>1 year) plaque psoriasis and a minimum body surface area involvement of 10% who were candidates for or had previously received systemic therapy or phototherapy. Each course consisted of once-weekly administration for 12 weeks (intravenous for Study 1, intramuscular for Study 2) of placebo or AMEVIVE®. Patients could receive concomitant low potency topical steroids. Concomitant phototherapy or systemic therapy was not allowed.
In Study 1, patients were randomized to receive one or two courses of AMEVIVE® 7.5 mg administered by intravenous bolus. The first and second courses in the two-course cohort were separated by at least a 12-week post-dosing interval. A total of 553 patients were randomized into three cohorts (Table 1).
| Course 1 (No. of patients) | Course 2 (No. of patients) | |
| Cohort 1 | AMEVIVE® (183) | AMEVIVE® (154) |
| Cohort 2 | AMEVIVE® (184) | Placebo (142) |
| Cohort 3 | Placebo (186) | AMEVIVE® (153) |
Study 2 provided a basis for comparison of patients treated with either 10 mg or 15 mg AMEVIVE® intramuscular. One hundred seventy-three patients were randomized to receive 10 mg of AMEVIVE® intramuscular, 166 to receive 15 mg of AMEVIVE® intramuscular, and 168 to receive placebo.
In Studies 1 and 2, 77% of patients had previously received systemic therapy and/or phototherapy for psoriasis. Of these, 23% and 19%, respectively, had failed to respond to at least one of these previous therapies.
Table 2 shows the treatment response in the first course of Study 1 and Study 2. Response to treatment in both studies was defined as the proportion of patients with a reduction in score on the Psoriasis Area and Severity Index (PASI) of at least 75% from baseline at two weeks following the 12-week treatment period.
Other treatment responses included the proportion of patients who achieved a scoring of “almost clear” or “clear” by Physician Global Assessment (PGA) and the proportion of patients with a reduction in PASI of at least 50% from baseline two weeks after the 12-week treatment period.
|
||||||
| Study 1 | Study 2 | |||||
|
Treatment response: (reduction in disease activity from baseline) |
Placebo (N=186) |
AMEVIVE® 7.5 mg intravenous (N=367)* | Difference (95% CI) |
Placebo (N=168) |
AMEVIVE® 15 mg intramuscular (N=166) | Difference (95% CI) |
| ≥75% reduction PASI | 4% | 14% | 10 (6, 15) | 5% | 21% | 16 (9, 23) |
| ≥50% reduction PASI | 10% | 38% | 28 (22, 35) | 18% | 42% | 24 (14, 33) |
| PGA “almost clear” or “clear” | 4% | 11% | 7 (3, 12) | 5% | 14% | 9 (3, 15) |
In Study 2, the proportion of responders to the 10 mg intramuscular dose was higher than placebo, but the difference was not statistically significant.
In both studies, onset of response to AMEVIVE® treatment (at least a 50% reduction of baseline PASI) began 60 days after the start of therapy.
With one course of therapy in Study 1 (intravenous route), the median duration of response (defined as maintenance of a 75% or greater reduction in PASI) was 3.5 months for AMEVIVE®-treated patients and 1 month for placebo-treated patients. In Study 2 (intramuscular route), the median duration of response was approximately 2 months for both AMEVIVE®-treated patients and placebo-treated patients.
Most patients who had responded to either AMEVIVE® or placebo maintained a 50% or greater reduction in PASI through the 3-month observation period.
Among responders in Study 1 who received AMEVIVE® 7.5 mg intravenous or in Study 2 who received AMEVIVE® 15 mg intramuscular and were followed off active treatment before AMEVIVE® retreatment, a 50% or greater reduction in PASI was maintained for a median of 7 months.
Some patients achieved their maximal response beyond 2 weeks post-dosing. In Studies 1 and 2, an additional 11% (42/367) and 7% (12/166) of patients treated with AMEVIVE®, respectively, achieved a 75% reduction from baseline PASI score at one or more visits after the first 2 weeks of the follow-up period.
Retreatment
Patients in Study 1 who had completed the first intravenous treatment course were eligible to receive a second treatment course if their psoriasis was less than “clear” by PGA and their CD4+ T lymphocyte count was above the lower limit of normal. The level of response (decrease in median PASI score) over the two courses of intravenous treatment is shown in Figure 1. The median reduction in PASI score was greater in patients who received a second course of AMEVIVE® treatment (see Cohort 1) compared to patients who received placebo (see Cohort 2).
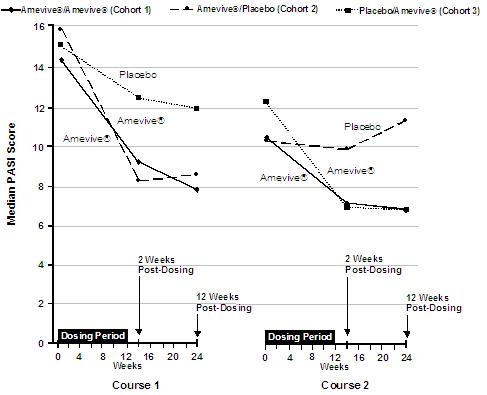
Figure 1. Median PASI Score Over Time
16 HOW SUPPLIED/STORAGE AND HANDLING
AMEVIVE® (alefacept) is a sterile, white to off-white, preservative-free lyophilizate (15 mg/vial) for intramuscular administration provided in single-use glass vials with a bromobutyl rubber stopper and an aluminum overseal. Each vial contains 15 mg of alefacept.
AMEVIVE® is available as follows:
| Carton Contents | NDC |
|
One- 5 mg single-use AMEVIVE® vial One - 10 mL single-use diluent vial of Sterile Water for Injection, USP One- 1 mL syringe Two- 23 gauge 1 ¼ inch needles | 0469-0021-04 |
|
Four -15 mg single-use AMEVIVE® vials Four -10 mL single-use diluent vials of Sterile Water for Injection, USP Four- 1 mL syringes Eight- 23 gauge 1 ¼ inch needles | 0469-0021-03 |
Store AMEVIVE® refrigerated between 2-8°C (36-46°F). Do not freeze. Store in carton until use to protect from light.
17 PATIENT COUNSELING INFORMATION
"See FDA-approved patient labeling (Medication Guide)"
Inform patients of the need for regular monitoring of white blood cell (lymphocyte) counts during therapy with AMEVIVE®. Inform patients that the reduction in lymphocytes could increase their chances of developing an infection or a malignancy. Advise patients to inform their physician promptly if they develop any signs of an infection or malignancy while undergoing a course of treatment with AMEVIVE®.
Advise female patients to notify their physicians if they become pregnant while taking AMEVIVE® or within 8 weeks of discontinuing AMEVIVE®.
Inform patients that serious liver injury has been reported in patients receiving AMEVIVE®. Patients should be advised to report to their physician persistent nausea, anorexia, fatigue, vomiting, abdominal pain, jaundice, easy bruising, dark urine or pale stools.
Inform patients that hypersensitivity reactions have been reported in patients receiving AMEVIVE®. Patients should promptly notify their physicians should urticaria or signs of angioedema (e.g. facial edema) develop.
AMEVIVE® (alefacept)
Manufactured by:
Astellas Pharma US, Inc.
Northbrook, IL 60062
US License # 1748
12E036-AMV
MEDICATION GUIDE
AMEVIVE® (Am-ah-veev)
(alefacept)for injection
Read this Medication Guide before your first treatment with AMEVIVE and if your doctor tells you that there is new information about AMEVIVE. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment.
What is the most important information I should know about AMEVIVE?
AMEVIVE can increase your risk of having serious side effects, including:
- Cancers: AMEVIVE may decrease the activity of your immune system and increase your risk for certain types of cancer. Before receiving AMEVIVE, tell your doctor if you have ever had any type of cancer.
- Serious Infections:
AMEVIVE lowers the number of certain white blood cells in your body. This can affect the ability of your immune system to fight infections and can increase your risk of getting serious infections. Some people get serious infections while receiving AMEVIVE, including infections caused by bacteria, fungi, or viruses. These infections may happen in your skin or a wound, lungs (pneumonia), stomach or intestine, gallbladder, or may spread throughout your body. Some people have to be treated in the hospital for their infection.
- Your doctor will do a blood test before you start receiving AMEVIVE and every 2 weeks during treatment to check your white blood cell counts.
- If your white blood cell counts are too low, your doctor will check your blood weekly. Your doctor may need to either delay or stop your treatment with AMEVIVE if your blood counts are too low.
You should not receive AMEVIVE if you have any kind of infection, unless your doctor says it is okay.
Before starting AMEVIVE, tell your doctor if you think you have an infection or have symptoms of an infection such as:
- swollen glands
- fever, sweats or chills
- cough
- muscle aches
- shortness of breath
- blood in your phlegm
- warm, red, or painful skin or sores on your body
- burning when you urinate or urinate more often than normal
- feel very tired
- get a lot of infections or have infections that keep coming back
After starting AMEVIVE, call your doctor right away if you have any symptoms of an infection (see above).
What is AMEVIVE?
AMEVIVE is a prescription medicine used to treat adults with moderate to severe plaque psoriasis that keeps coming back (chronic), who may benefit from taking injections or pills (systemic therapy) or phototherapy (treatment using ultraviolet light alone or with pills).
It is not known if AMEVIVE is safe and effective in children.
Who should not use AMEVIVE?
Do not use AMEVIVE if you:
- have HIV infection
What should I tell my doctor before taking AMEVIVE?
Before receiving AMEVIVE, tell your doctor if you:
- have any of the conditions or symptoms listed in the section “What is the most important information I should know about AMEVIVE?”
- are receiving phototherapy for your psoriasis.
- are allergic to AMEVIVE or any of its ingredients. See the end of this Medication Guide for a list of the ingredients in AMEVIVE.
- have any other medical conditions.
- are pregnant or plan to become pregnant. It is not known if AMEVIVE will harm your unborn baby. Tell your doctor right away if you become pregnant while receiving AMEVIVE or within 8 weeks after receiving your last dose of AMEVIVE.
- are breastfeeding or plan to breastfeed. It is not known if AMEVIVE passes into your breast milk. You and your doctor should decide if you will take AMEVIVE or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Especially tell your doctor if you receive phototherapy or take any other medicines that affect your immune system, including other medicines for treatment of your plaque psoriasis.
How will I receive AMEVIVE?
- AMEVIVE is given as an injection into your muscle.
- AMEVIVE is usually given one time each week for 12 weeks.
- Talk with your doctor to find out when you will receive injections. Keep all of your injection and follow-up appointments. It is important for you to stay under your doctor’s care during treatment.
• After you finish 12 weeks of treatment, you will stop treatment for at least 12 weeks.
Your doctor may then decide that you should receive another 12 week treatment course of AMEVIVE.
What are the possible side effects of AMEVIVE?
AMEVIVE can cause serious side effects, including:
- See “What is the most important information I should know about AMEVIVE?”
- Serious allergic reactions can happen during treatment with AMEVIVE.
Call your doctor right away if you get any of the following symptoms of an allergic reaction with AMEVIVE:
-
- red-itchy welts (hives) on your skin
- swelling or tingling of your lips, tongue, or inside your mouth
- Serious Liver problems. Some patients treated with AMEVIVE develop serious liver problems, including liver injury.
Call your doctor right away if you get any of the following signs and symptoms of serious liver problems:
-
- yellowing of your skin or the whites of your eyes
- you start to bruise easily
- tiredness
- loss of appetite or vomiting that does not go away, or abdominal pain
- dark urine
- pale stool
Common side effects of AMEVIVE include:
- sore throat
- dizziness
- cough
- nausea
- itching
- muscle aches
- chills
- injection site reactions such as pain, redness
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of AMEVIVE. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about AMEVIVE.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide.
This Medication Guide summarizes the most important information about AMEVIVE. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about AMEVIVE that was written for healthcare professionals.
What are the ingredients in AMEVIVE?
Active ingredient: alefacept
Inactive Ingredients: citric acid monohydrate, glycine, sodium citrate dehydrate, sucrose. Sterile water for injection is provided as a diluent.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Astellas Pharma US, Inc.
Northbrook, IL 60062
Issued May 2012
12E036-AMV-MG
| AMEVIVE
alefacept kit |
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
|
|||||||||||||||||||||
| Marketing Information | |||
| Marketing Category | Application Number or Monograph Citation | Marketing Start Date | Marketing End Date |
| BLA | BLA125036 | 01/30/2003 | 09/28/2012 |
| Labeler - Astellas Pharma US Inc. (605764828) |
Revised: 05/2012 Astellas Pharma US Inc.