Label: PARICALCITOL capsule, liquid filled



- NDC Code(s): 49483-687-03, 49483-688-03, 49483-689-03
- Packager: TIME CAP LABORATORIES, INC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated December 2, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use
PARICALCITOL CAPSULES safely and effectively.
See Full Prescribing Information for PARICALCITOL CAPSULES.
PARICALCITOL capsules, for oral use
Initial U.S. Approval:1998INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Initial Dosage:CKD Stage 3 and 4 (2.1) Adult:Baseline iPTH ≤ 500 pg/mL 1 mcg orally daily or 2 mcg three times a week* Adult: Baseline iPTH > 500 pg/mL 2 mcg orally daily or 4 mcg three times a week* Dose Titration : CKD Stage 3 and 4 (2.1) Adult : iPTH same, increased or decreased by <30% relative to baseline Increase dose by 1 mcg daily or 2 mcg three times a week* Adult : iPTH decreased by ≥ 30% and ≤ 60% relative to baseline Maintain dose Adult : iPTH decreased by > 60% or iPTH < 60% pg/mL relative to baseline Decrease dose by 1 mcg daily or 2 mcg three times a week*
*Not more frequently than every other day when dosing three times a week
(2)Initial dosage : CKD Stage 5 (2.2)
Adult
Dose (micrograms) = baseline iPTH (pg/mL) divided by 80. Administer dose orally three times a week*.
Dose Titration: CKD Stage 5 (2.2)
Adult
Dose in micrograms is based on most recent iPTH (pg/mL) divided by 80 with adjustments based on serum calcium and phosphorous levels. Dose three times a week*
* Not more frequently than every other day. (2)
- CKD Stage 5: To avoid hypercalcemia only treat patients after their baseline serum calcium has been reduced to 9.5 mg/dL or lower (2.2).
DOSAGE FORMS AND STRENGTHS
Capsules: 1 mcg, 2 mcg, and 4 mcg (3). (3)
CONTRAINDICATIONS
- Evidence of hypercalcemia (4).
- Evidence of vitamin D toxicity (4).
WARNINGS AND PRECAUTIONS
- Hypercalcemia: Excessive administration of Paricalcitol Capsules can cause over suppression of PTH, hypercalcemia, hypercalciuria, hyperphosphatemia, and adynamic bone disease. Prescription-based doses of vitamin D and its derivatives should be withheld during Paricalcitol treatment (5.1).
- Digitalis toxicity: Potentiated by hypercalcemia of any cause. Use caution when Paricalcitol Capsules are prescribed concomitantly with digitalis compounds (5.2).
- Laboratory tests: Monitor serum calcium, serum phosphorus, and serum or plasma iPTH during initial dosing or following any dose adjustment. Paricalcitol Capsules may increase serum creatinine and therefore decrease the estimated GFR (eGFR) (5.3).
- Aluminum overload and toxicity: Avoid excessive use of aluminum containing compounds (5.4).
ADVERSE REACTIONS
The most common adverse reactions (> 5% and more frequent than placebo) include diarrhea, nasopharyngitis, dizziness, vomiting, hypertension, hypersensitity, nausea, and edema (6). (6)
(6)
(6)
To report SUSPECTED ADVERSE REACTIONS, contact Marksans at 1-877-376-4271 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch (6)
(6)
(6)
DRUG INTERACTIONS
- Strong CYP3A inhibitors (e.g. ketoconazole) will increase the exposure of paricalcitol. Use with caution (7).
- Cholestyramine, Mineral Oil: Intestinal absorption of paricalcitol may be reduced if administered simultaneously with cholestyramine or mineral oil. Take paricalcitol capsules at least one hour before or 4 to 6 hours after taking cholestyramine or mineral oil (7).
USE IN SPECIFIC POPULATIONS
Lactation: Breastfeeding not recommended (8.2). (8)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS & USAGE
2 DOSAGE & ADMINISTRATION
3 DOSAGE FORMS & STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
6 ADVERSE REACTIONS
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS & USAGE
1.1 Chronic Kidney Disease Stages 3 and 4
Paricalcitol Capsules are indicated in adults for the prevention and treatment of secondary hyperparathyroidism associated with Chronic Kidney Disease (CKD) Stages 3 and 4.
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
1.2 Chronic Kidney Disease Stage 5
Paricalcitol Capsules are indicated in adults for the prevention and treatment of secondary hyperparathyroidism associated with CKD Stage 5 in patients on hemodialysis (HD) or peritoneal dialysis (PD).
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
-
2 DOSAGE & ADMINISTRATION
2.1 Chronic Kidney Disease Stages 3 and 4 in Adults
Administer Paricalcitol Capsules orally once daily or three times a week. [see Clinical Studies(14.1)] . When dosing three times weekly, do not administer more frequently than every other day.
Initial Dose
Table 1. Recommended Paricalcitol Capsules Starting Dose Based upon Baseline iPTH Level
Baseline iPTH Level
Daily Dose
Three Times a Week Dose*
Less than or equal to 500 pg/mL
1 mcg
2 mcg
More than 500 pg/mL
2 mcg
4 mcg
* To be administered not more often than every other day
Dose Titration
Table 2. Recommended Paricalcitol Capsules Dose Titration Base upon iPTH Level
Dose Adjustment at 2 to 4
Week Intervals
iPTH Level Relative to Baseline
Paricalcitol
Capsule Dose
Daily Dosage
Three Times a
Week Dosage*
The same, increased or decreased by less than 30%
Increase dose by
1 mcg
2 mcg
Decreased by more than or equal to 30% and less than or equal to 60%
Maintain dose
-
-
Decreased by more than 60%
or iPTH less than 60 pg/mL
Decrease dose
by
1 mcg
2 mcg
* To be administered not more often than every other day
If a patient is taking the lowest dose, 1 mcg, on the daily regimen and a dose reduction is needed, the dose can be decreased to 1 mcg three times a week. If a further dose reduction is required, the drug should be withheld as needed and restarted at a lower dosing frequency.
2.2 Chronic Kidney Disease Stage 5 in Adults
Initial Dose
Administer the dose of Paricalcitol Capsules orally three times a week, no more frequently than every other day upon the following formula: Dose (micrograms) = baseline iPTH (pg/mL) divided by 80. Treat patients only after their baseline serum calcium has been adjusted to 9.5 mg/dL or lower to minimize the risk of hypercalcemia [see Clinical Pharmacology (12.2) and Clinical Studies (14.2)].
Dose Titration
Individualize the dose of Paricalcitol Capsules based on iPTH, serum calcium and phosphorus level. Titrate Paricalcitol Capsules dose based on following formula: Dose (micrograms) = most recent iPTH level (pg/ml) divided by 80 If serum calcium is elevated, the dose should be decreased by 2 to 4 micrograms.
As iPTH approaches the target range, small, individualized dose adjustments may be necessary in order to achieve a stable iPTH. In situations where monitoring of iPTH, Ca or P occurs less frequently than once per week, a more modest initial and dose titration ratio ((e.g., iPTH divided by 100) may be warranted.
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
2.4 Monitoring
Monitor serum calcium and phosphorus levels closely after initiation of Paricalcitol Capsules during dose titration periods and during co-administration with strong CYP3A inhibitors [see Warnings and Precautions(5.3), Drug Interactions (7), and Clinical Pharmacology (12.3)].
If hypercalcemia is observed, the dose of Paricalcitol Capsules should be reduced or withheld until these parameters are normalized.
2.5 Administration
Paricalcitol Capsules may be taken without regard to food.
-
3 DOSAGE FORMS & STRENGTHS
Paricalcitol Capsules are available as 1 mcg, 2 mcg, and 4 mcg soft gelatin capsules.
- 1 mcg-grey colour oval shaped soft gelatin capsules imprinted with "12" in black ink.
- 2 mcg-brown colour oval shaped soft gelatin capsules imprinted with "14" in black ink.
- 4 mcg-light yellow colour oval shaped soft gelatin capsules imprinted with "17" in black ink.
-
4 CONTRAINDICATIONS
Paricalcitol Capsules should not be given to patients with evidence of
- hypercalcemia or
- vitamin D toxicity [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
Excessive administration of vitamin D compounds. including Paricalcitol Capsules, can cause over suppression of PTH, hypercalcemia, hypercalciuria, hyperphosphatemia, and adynamic bone disease.
5.1 HypercalcemiaProgressive hypercalcemia due to overdosage of vitamin D and its metabolites may be so severe as to require emergency attention [ see Overdosage (10)] . Acute hypercalcemia may exacerbate tendencies for cardiac arrhythmias and seizures and may potentiate the action of digitalis. Chronic hypercalcemia can lead to generalized vascular calcification and other soft-tissue calcification. Concomitant administration of high doses of calcium-containing preparations or thiazide diueretics with Paricalcitol may increase the risk of hypercalcemia. High intake of calcium and phosphate concomitant with vitamin D compounds may lead to serum abnormalities requiring more frequent patient monitoring and individualized dose titration. Patients also should be informed about the symptoms of elevated calcium, which include feeling tired, difficulty thinking clearly, loss of appetite, nausea, vomiting, constipation, increased thirst, increased urination and weight loss.
Prescription-based doses of vitamin D and its derivatives should be withheld during Paricalcitol treatment to avoid hypercalcemia.5.2 Digitalis Toxicity
Digitalis toxicity is potentiated by hypercalcemia of any cause. Use caution when Paricalcitol Capsules are prescribed concomitantly with digitalis compounds.
5.3 Laboratory Tests
During the initial dosing or following any dose adjustment of medication, serum calcium, serum phosphorus, and serum or plasma iPTH should be monitored at least every two weeks for 3 months, then monthly for 3 months, and every 3 months thereafter.
In pre-dialysis patients, Paricalcitol Capsules may increase serum creatinine and therefore decrease the estimated GFR (eGFR). Similar effects have also been seen with calcitriol.
5.4 Aluminum Overload and Toxicity
Aluminum-containing preparations (e.g., antacids, phosphate binders) should not be administered chronically with Paricalcitol, as increased blood levels of aluminum and aluminum bone toxicity may occur.
-
6 ADVERSE REACTIONS
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
CKD Stages 3 and 4
Adults
The safety of Paricalcitol Capsules has been evaluated in three 24-week (approximately six-month), double-blind, placebo-controlled, multicenter clinical studies involving 220 CKD Stages 3 and 4 patients. Six percent (6%) of Paricalcitol Capsules treated patients and 4% of placebo treated patients discontinued from clinical studies due to an adverse event. Adverse events occurring in the Paricalcitol Capsules group at a frequency of 2% or greater and more frequently than in the placebo group are presented in: Table 3.
Table 3. Adverse Reactions by Body System Occurring in ≥ 2% of Subjects in the Paricalcitol Capsules-Treated Group of Three, Double-Blind, Placebo-Controlled CKD Stages 3 and 4 Studies
Number (%) of Subjects
Adverse Events a
Paricalcitol Capsules
(n = 107)
Placebo (n =113)
Overall
88
(82%)
86
(76%)
Ear and Labyrinth Disorders
Vertigo
5
(5%)
0
(0%)
Gastrointestinal Disorders
Abdominal Discomfort
4
(4%)
1
(1%)
Constipation
4
(4%)
4
(4%)
Diarrhea
7
(7%)
5
(4%)
Nausea
6
(6%)
4
(4%)
Vomiting
5
(5%)
5
(4%)
General Disorders and Administration Site Conditions
Chest Pain
3
(3%)
1
(1%)
Edema
6
(6%)
5
(4%)
Pain
4
(4%)
4
(4%)
Immune System Disorders
Hypersensitivity
6
(6%)
2
(2%)
Infections and Infestations
Fungal Infection
3
(3%)
0
(0%)
Gastroenteritis
3
(3%)
3
(3%)
Infection
3
(3%)
3
(3%)
Sinusitis
3
(3%)
1
(1%)
Urinary Tract Infection
3
(3%)
1
(1%)
Viral Infection
8
(7%)
8
(7%)
Metabolism and Nutrition Disorders
Dehydration
3
(3%) 1 (1%) Musculoskeletal and Connective Tissue Disorders
Arthritis
5
(5%)
0
(0%)
Back Pain
3
(3%)
1
(1%)
Muscle Spasms
3
(3%)
0
(0%)
Nervous System Disorders
Dizziness
5
(5%)
5
(4%)
Headache
5
(5%)
5
(4%)
Syncope
3
(3%)
1
(1%)
Psychiatric Disorders
Depression
3
(3%)
0
(0%)
Respiratory, Thoracic and Mediastinal Disorders
Cough
3
(3%)
2
(2%)
Oropharyngeal Pain
4
(4%)
0
(0%)
Skin and Subcutaneous Tissue Disorders
Pruritus
3
(3%)
3
(3%)
Rash
4
(4%)
1
(1%)
Skin Ulcer
3
(3%)
0
(0%)
Vascular Disorders
Hypertension
7
(7%)
4
(4%)
Hypotension
5
(5%)
3
(3%)
a. Includes only events more common in the Paricalcitol treatment group.
Additional Adverse Reactions
The following additional adverse reactions , occurred in <2% of the Paricalcitol-treated adult patients in the above double-blind, placebo-controlled clinical trial.
Gastrointestinal Disorders: Dry mouth
Investigations: Hepatic enzyme abnormal
Nervous System Disorders: Dysgeusia
Skin and Subcutaneous Tissue Disorders: Urticaria
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
CKD Stage 5
Adults
The safety of Paricalcitol Capsules has been evaluated in one 12-week, double-blind, placebo controlled, multicenter clinical study involving 88 CKD Stage 5 patients. Sixty-one patients received Paricalcitol Capsules and 27 patients received placebo.
The proportion of patients who terminated prematurely from the study due to adverse events was 7% for Paricalcitol Capsules treated patients and 7% for placebo patients.
Adverse events occurring in the Paricalcitol Capsules group at a frequency of 2% or greater and more frequently than in the placebo group are as follows:
Table 4. Adverse Reactions by Body System Occurring in ≥ 2% of Subjects in the Paricalcitol Capsules-Treated Group, Double-Blind, Placebo-Controlled CKD Stage 5 Study
Number (%) of Subjects
Adverse Events a
Paricalcitol Capsules
(n = 61)
Placebo (n =27)
Overall
43
(70%)
19
(70%)
Gastrointestinal Disorders
Constipation
3
(5%)
0
(0%)
Diarrhea
7
(11%)
3
(11%)
Vomiting
4
(7%)
0
(0%)
General Disorders and Administration Site Conditions
Fatigue
2
(3%)
0
(0%)
Edema peripheral
2
(3%)
0
(0%)
Infections and Infestations
Nasopharyngitis
5
(8%)
2
(7%)
Peritonitis
3
(5%)
0
(0%)
Sinusitis
2
(3%)
0
(0%)
Urinary Tract Infection
2
(3%)
0
0%)
Metabolism and Nutrition Disorders
Fluid overload
3
(5%)
0
(0%)
Hypoglycemia
2
(3%)
0
(0%)
Nervous System Disorders
Dizziness
4
(7%)
0
( 0%)
Headache
2
(3%)
0
(0%)
Psychiatric Disorders
Anxiety
2
(3%)
0
(0%)
Insomnia
3
(5%)
0
(0%)
Renal and Urinary Disorders
Renal failure Chronic
2
(3%)
0
(0%)
a. Includes only events more common in the Paricalcitol treatment group.
Additional Adverse Reactions
The following adverse reactions, occurred in <2% of the Paricalcitol Capsules- treated patients in the above double-blind, placebo-controlled clinical trial.
Gastrointestinal Disorders: Gastroesophageal reflux disease
Metabolism and Nutrition Disorders: Decreased appetite, hypercalcemia, hypocalcemia
Reproductive System and Breast Disorders: Breast tendernessSkin and Subcutaneous Tissue Disorders: Acne
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
6.2 Postmarketing Experience
The following additional adverse reactions have been reported during post-approval use of Paricalcitol Capsules. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a casual relationship to drug exposure.
Immune System Disorders: Angioedema (including laryngeal edema)Investigations: Blood creatinine increased
-
7 DRUG INTERACTIONS
Table 5 shows the clinically significant drug interactions with Paricalcitol capsules.
Table 5: Clinically Significant Drug Interactions with Paricalcitol
CYP3A Inhibitors
Clinical Impact
Paricalcitol is partially metabolized by CYP3A. Hence, exposure of paricalcitol will increase upon coadministration with strong CYP3A inhibitors such as but not limited to: boceprevir, clarithromycin, conivaptan, grapefruit juice, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, mibefradil, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telaprevir, telithromycin, voriconazole.
Intervention
Dose adjustment of Paricalcitol capsules may be necessary. Monitor closely for iPTH and serum calcium concentrations, if a patient initiates or discontinues therapy with a strong CYP3A4 inhibitor.
Cholestyramine
Clinical Impact
Drugs that impair intestinal absorption of fat-soluble vitamins, such as cholestyramine, may interfere with the absorption of paricalcitol.
Intervention
Recommend to take Paricalcitol capsules at least 1 hour before or 4 to 6 hours after taking cholestyramine (or at as great an interval as possible) to avoid impeding absorption of paricalcitol.
Mineral Oil
Clinical Impact
Mineral oil or other substances that may affect absorption of fat may influence the absorption of paricalcitol.
Intervention
Recommend to take Paricalcitol capsules at least 1 hour before or 4 to 6 hours after taking mineral oil (or at as great an interval as possible) to avoid affecting absorption of paricalcitol.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited data with Paricalcitol capsules in pregnant women are insufficient to inform a drug- associated risk for major birth defects and miscarriage. There are risks to the mother and fetus associated with chronic kidney disease in pregnancy [see Clinical Considerations].
In animal reproduction studies, slightly increased embryofetal loss was observed in pregnant rats and rabbits administered paricalcitol intravenously during the period of organogenesis at doses 2 and 0.5 times, respectively, the maximum recommended human dose (MRHD). Adverse reproductive outcomes were observed at doses that caused maternal toxicity [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other
adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal riskChronic kidney disease in pregnancy increases the maternal risk for hypertension, spontaneous abortion, preterm labor, and preeclampsia. Chronic kidney disease increases the fetal risk for intrauterine growth restriction (IUGR), prematurity, polyhydramnios, still birth, and low birth weight.
Data
Animal Data
Pregnant rats and rabbits were treated with paricalcitol by once-daily intravenous (IV) injection during the period of organogenesis (in rats, from gestation day (GD) 6 to 17; in rabbits, from GD 6 to 18). Rats were dosed at 0, 0.3, 1.0 or 3.0 mcg/kg/day and rabbits at 0, 0.03, 0.1 or 0.3 mcg/kg/day, representing up to 2 or 0.5 times, respectively, the maximum recommended human dose (MRHD) of 0.24 mcg/kg, based on body surface area (mg/m 2 ). Slightly decreased fetal viability was observed in both studies at the highest doses representing 2 and 0.5 times, respectively, the MRHD in the presence of maternal toxicity (decreased body weight and food consumption). Pregnant rats were administered paricalcitol by IV injection three times per week at doses of 0, 0.3, 3.0 or 20.0 mcg/kg/day throughout gestation, parturition and lactation (GD 6 to lactation day (LD) 20) representing exposures up to 13 times the MHRD. A small increase in stillbirths and pup deaths from parturition to LD 4 were observed at the high dose when compared to the control group (9.2% versus 3.3% in controls) at 13 times the MRHD, which occurred at a maternally toxic dose known to cause hypercalcemia in rats. Surviving pups were not adversely affected; body weight gains, developmental landmarks, reflex ontogeny, learning indices, and locomotor activity were all within normal parameters. F1 reproductive capacity was unaffected.
8.2 Lactation
Risk Summary
There is no information available on the presence of paricalcitol in human milk, the effects of the drug on the breastfed infant or the effects of the drug on milk production. Studies in rats have shown that paricalcitol and/or its metabolites are present in the milk of lactating rats; however, due to specifies-specific differences in lactation physiology, animal data may not reliably predict drug levels in human milk [see Data]. Because of the potential for serious adverse reactions, including hypercalcemia in a breastfed infant, advise patients that breastfeeding is not recommended during treatment with Paricalcitol.
Data
Following a single oral administration of 20 mcg/kg of radioactive [3H] paricalcitol to lactating rats, the concentrations of total radioactivity was determined. Lower levels of total radioactivity were present in the milk compared to that in the plasma of the dams indicating that low levels of [3H] paricalcitol and/or its metabolites are secreted into milk. Exposure of the pups to[3H] paricalcitol through milk was confirmed by the presence of radioactive material in the pups’ stomachs.
8.4 Pediatric Use
Safety and effectiveness of Paricalcitol Capsules in pediatric patients under the age of 10 years have not been established.
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
8.5 Geriatric Use
Of the total number (n = 220) of CKD Stages 3 and 4 patients in clinical studies of Paricalcitol capsules, 49% were age 65 and over, while 17% were age 75 and over. Of the total number (n =88) of CKD Stage 5 patients in the pivotal study of Paricalcitol Capsules, 28% were age 65 and over, while 6% were age 75 and over. No overall differences in safety and effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Excessive administration of Paricalcitol Capsules can cause hypercalcemia, hypercalciuria, and hyperphosphatemia, and over suppression of PTH [ see Warnings and Precautions(5.1) ].
Treatment of Overdosage
The treatment of acute overdosage of Paricalcitol Capsules should consist of general supportive measures. If drug ingestion is discovered within a relatively short time, induction of emesis or gastric lavage may be of benefit in preventing further absorption. If the drug has passed through the stomach, the administration of mineral oil may promote its fecal elimination. Serial serum electrolyte determinations (especially calcium), rate of urinary calcium excretion, and assessment of electrocardiographic abnormalities due to hypercalcemia should be obtained. Such monitoring is critical in patients receiving digitalis. Discontinuation of supplemental calcium and institution of a low-calcium diet are also indicated in accidental overdosage. Due to the relatively short duration of the pharmacological action of paricalcitol, further measures are probably unnecessary. If persistent and markedly elevated serum calcium levels occur, there are a variety of therapeutic alternatives that may be considered depending on the patient's underlying condition. These include the use of drugs such as phosphates and corticosteroids, as well as measures to induce an appropriate forced diuresis.
Paricalcitol is not significantly removed by dialysis. -
11 DESCRIPTION
Paricalcitol, USP, the active ingredient in Paricalcitol Capsules, is a synthetically manufactured, metabolically active vitamin D analog of calcitriol with modifications to the side chain (D 2) and the A (19-nor) ring. Paricalcitol is available as soft gelatin capsules for oral administration containing 1 microgram, 2 micrograms, and 4 micrograms of paricalcitol. Each capsule also contains medium chain triglycerides, alcohol, and butylated hydroxytoluene. ..The capsule shell is composed of gelatin, glycerin, Noncrystallizing Sorbitol solution, titanium dioxide, iron oxide red (2 microgram capsules only), iron oxide yellow (2 microgram and 4 microgram capsules), iron oxide black (1 microgram capsules only) and water. The soft gelatin capsules are printed with black ink Opacode Black (S-1-17823) containing Isopropyl alcohol, Black iron oxide, N-Butyl alcohol, Propylene glycol, Ammonium hydroxide and Shellac.
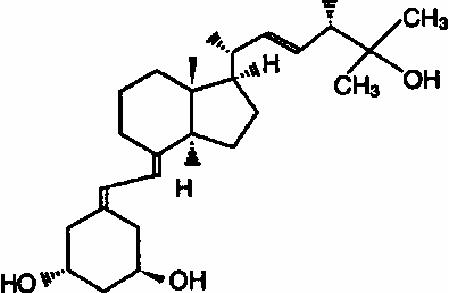
Paricalcitol is a white, crystalline powder with the empirical formula of C 27H 44O 3, which corresponds to a molecular weight of 416.64. Paricalcitol is chemically designated as 19-nor-1α,3β,25-trihydroxy-9,10-secoergosta-5(Z),7(E),22(E)- triene and has the following structural formula:
-
12 CLINICAL PHARMACOLOGY
Secondary hyperparathyroidism is characterized by an elevation in parathyroid hormone (PTH) associated with inadequate levels of active vitamin D hormone. The source of vitamin D in the body is from synthesis in the skin as vitamin D 3 and from dietary intake as either vitamin D 2 or D 3. Both vitamin D 2 and D 3 require two sequential hydroxylations in the liver and the kidney to bind to and to activate the vitamin D receptor (VDR). The endogenous VDR activator, calcitriol [1,25(OH) 2D 3], is a hormone that binds to VDRs that are present in the parathyroid gland, intestine, kidney, and bone to maintain parathyroid function and calcium and phosphorus homeostasis, and to VDRs found in many other tissues, including prostate, endothelium and immune cells. VDR activation is essential for the proper formation and maintenance of normal bone. In the diseased kidney, the activation of vitamin D is diminished, resulting in a rise of PTH, subsequently leading to secondary hyperparathyroidism and disturbances in the calcium and phosphorus homeostasis. Decreased levels of 1,25(OH) 2D 3 have been observed in early stages of chronic kidney disease. The decreased levels of 1,25(OH) 2D 3 and resultant elevated PTH levels, both of which often precede abnormalities in serum calcium and phosphorus, affect bone turnover rate and may result in renal osteodystrophy.
12.1 Mechanism of Action
Paricalcitol is a synthetic, biologically active vitamin D 2 analog of calcitriol. Preclinical and in vitro studies have demonstrated that paricalcitol's biological actions are mediated through binding of the VDR, which results in the selective activation of vitamin D responsive pathways. Vitamin D and paricalcitol have been shown to reduce parathyroid hormone levels by inhibiting PTH synthesis and secretion.
12.2 Pharmacodynamics
12.2 Pharmacodynamics
Paricalcitol decreases serum intact parathyroid hormone (iPTH) and increases serum calcium and serum phosphorous in both HD and PD patients. This observed relationship was quantified using a mathematical model for HD and PD patient populations separately. Computer-based simulations of 100 trials in HD or PD patients (N = 100) using these relationships predict slightly lower efficacy (at least two consecutive ≥ 30% reductions from baseline iPTH) with lower hypercalcemia rates (at least two consecutive serum calcium ≥ 10.5 mg/dL) for lower iPTH-based dosing regimens. Further lowering of hypercalcemia rates was predicted if the treatment with paricalcitol is initiated in patients with lower serum calcium levels at screening.
Based on these simulations, a dosing regimen of iPTH/80 with a screening serum calcium ≤ 9.5 mg/dL, approximately 76.5% (95% CI: 75.6% – 77.3%) of HD patients are predicted to achieve at least two consecutive weekly ≥ 30% reductions from baseline iPTH over a duration of 12 weeks. The predicted incidence of hypercalcemia is 0.8% (95% CI: 0.7% – 1.0%). In PD patients, with this dosing regimen, approximately 83.3% (95% CI: 82.6% – 84.0%) of patients are predicted to achieve at least two consecutive weekly ≥ 30% reductions from baseline iPTH. The predicted incidence of hypercalcemia is 12.4% (95% CI: 11.7% -13.0%) [see Clinical Studies (14.2) and Dosage and Administration (2.2) ].
12.3 Pharmacokinetics
Absorption
The mean absolute bioavailability of Paricalcitol Capsules under low-fat fed condition ranged from 72% to 86% in healthy adult volunteers, CKD Stage 5 patients on HD, and CKD Stage 5 patients on PD. A food effect study in healthy adult volunteers indicated that the C max and AUC 0-∞, were unchanged when paricalcitol was administered with a high fat meal compared to fasting. Food delayed T max by about 2 hours. The AUC 0-∞ of paricalcitol increased proportionally over the dose range of 0.06 to 0.48 mcg/kg in healthy adult volunteers.
Distribution
Paricalcitol is extensively bound to plasma proteins (≥ 99.8%). The mean apparent volume of distribution following a 0.24 mcg/kg dose of paricalcitol in healthy adult volunteers was 34 L. The mean apparent volume of distribution following a 4 mcg dose of paricalcitol in CKD Stage 3 and a 3 mcg dose in CKD Stage 4 patients is between 44 and 46 L.
Metabolism
After oral administration of a 0.48 mcg/kg dose of 3H-paricalcitol, parent drug was extensively metabolized, with only about 2% of the dose eliminated unchanged in the feces, and no parent drug was found in the urine. Several metabolites were detected in both the urine and feces. Most of the systemic exposure was from the parent drug. Two minor metabolites, relative to paricalcitol, were detected in human plasma. One metabolite was identified as 24(R)-hydroxy paricalcitol, while the other metabolite was unidentified. The 24(R)-hydroxy paricalcitol is less active than Paricalcitol in an in vivo rat model of PTH suppression.
In vitro data suggest that paricalcitol is metabolized by multiple hepatic and non-hepatic enzymes, including mitochondrial CYP24, as well as CYP3A4 and UGT1A4. The identified metabolites include the product of 24(R)-hydroxylation, 24,26- and 24,28-dihydroxylation and direct glucuronidation.
Elimination
Paricalcitol is eliminated primarily via hepatobiliary excretion; approximately 70% of the radiolabeled dose is recovered in the feces and 18% is recovered in the urine. While the mean elimination half-life of paricalcitol is 4 to 6 hours in healthy adult volunteers,the mean elimination half-life of paricalcitol in CKD Stages 3,4, and 5 (on HD and PD) patients ranged from 14 to 20 hours.
Table 6. Paricalcitol Capsule Pharmacokinetic Parameters(mean + SD) in CKD Stages 3, 4, and 5 Adult Patients
Pharmacokinetic Parameters(units)
CKD Stage 3
n = 15*
CKD Stage 4
n = 14*
CKD Stage 5 HD**
n=14
CKD Stage 5 PD**
n=8
C max (ng/mL)
0.11 ± 0.04
0.06 ±0.01
0.575 ± 0.17
0.413 ± 0.06
AUC 0-∞ (ng•h/mL)
2.42 ± 0.61
2.13 ± 0.73
11.67 ± 3.23
13.41 ± 5.48
CL/F(L/h)
1.77 ±0.50
1.52 ± 0.36
1.82 ± 0.75
1.76 ± 0.77
V/F (L)
43.7 ±14.4
46.4 ±12.4
38 ± 16.4
48.7 ± 15.6
t 1/2
16.8 ± 2.65
19.7 ±7.2
13.9 ± 5.1
17.7 ± 9.6
HD:hemodialysis;PD:peritoneal dialysis.*Four mcg Paricalcitol capsules were given to CKD Stage 3 patients; three mcg Paricalcitol capsules were given to CKD Stage 4 patients.
**CKD Stage 5 HD and PD patients received a 0.24 mcg/kg dose of paricalcitol as capsules.
Specific Populations
Geriatric
The pharmacokinetics of paricalcitol has not been investigated in geriatric patients greater than 65 years [see Use in Specific Populations (8.5)] .
Pediatric
Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
Gender
The pharmacokinetics of paricalcitol following single doses over the 0.06 to 0.48 mcg/kg dose range was gender independent.
Hepatic Impairment
The disposition of paricalcitol (0.24 mcg/kg) was compared in patients with mild (n = 5) and moderate (n = 5) hepatic impairment (as indicated by the Child-Pugh method) and subjects with normal hepatic function (n = 10). The pharmacokinetics of unbound paricalcitol was similar across the range of hepatic function evaluated in this study. No dose adjustment is required in patients with mild and moderate hepatic impairment. The influence of severe hepatic impairment on the pharmacokinetics of paricalcitol has not been evaluated.
Renal ImpairmentFollowing administration of paricalcitol capsules, the pharmacokinetic profile of paricalcitol for CKD Stage 5 on HD or PD was comparable to that in CKD 3 or 4 patients. Therefore, no special dose adjustments are required other than those recommended in the Dosage and Administration section [see Dosage and Administration (2)].
Drug Interactions
An in vitro study indicates that paricalcitol is neither an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 or CYP3A nor an inducer of CYP2B6, CYP2C9 or CYP3A. Hence, paricalcitol is neither expected to inhibit nor induce the clearance of drugs metabolized by these enzymes.
Omeprazole
The effect of omeprazole (40 mg capsule), a strong inhibitor of CYP2C19, on paricalcitol (four 4 mcg capsules) pharmacokinetics was investigated in a single dose, crossover study in healthy subjects. The pharmacokinetics of paricalcitol was not affected when omeprazole was administered approximately 2 hours prior to the paricalcitol dose.
Ketoconazole
The effect of multiple doses of ketoconazole, a strong inhibitor of CYP3A, administered as 200 mg BID for 5 days on the pharmacokinetics of paricalcitol (4 mcg capsule) has been studied in healthy subjects. The C max of paricalcitol was minimally affected, but AUC o-∞. approximately doubled in the presence of ketoconazole. The mean half-life of paricalcitol was 17.0 hours in the presence of ketoconazole as compared to 9.8 hours, when paricalcitol was administered alone [see Drug Interactions (7)]. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
13.1 Carcinogenesis, Mutagenesis and Impairment of Fertility
In a 104-week carcinogenicity study in CD-1 mice, an increased incidence of uterine leiomyoma and leiomyosarcoma was observed at subcutaneous doses of 1, 3,10 mcg/kg given three times weekly (2 to 15 times the AUC at a human dose of 14 mcg, equivalent to 0.24 mcg/kg based on AUC). The incidence rate of uterine leiomyoma was significantly different than the control group at the highest dose of 10 mcg/kg. In a 104-week carcinogenicity study in rats, there was an increased incidence of benign adrenal pheochromocytoma at subcutaneous doses of 0.15, 0.5, 1.5 mcg/kg (< 1 to 7 times the exposure following a human dose of 14 mcg, equivalent to 0.24 mcg/kg based on AUC). The increased incidence of pheochromocytomas in rats may be related to the alteration of calcium homeostasis by paricalcitol. Paricalcitol did not exhibit genetic toxicity in vitro with or without metabolic activation in the microbial mutagenesis assay (Ames Assay), mouse lymphoma mutagenesis assay (L5178Y), or a human lymphocyte cell chromosomal aberration assay. There was also no evidence of genetic toxicity in an in vivo mouse micronucleus assay. Paricalcitol had no effect on fertility (male or female) in rats at intravenous doses up to 20 mcg/kg/dose (equivalent to 13 times a human dose of 14 mcg based on surface area, mcg/m 2).
-
14 CLINICAL STUDIES
14.1 Chronic Kidney Disease Stages 3 and 4
AdultsThe safety and efficacy of Paricalcitol Capsules were evaluated in three, 24-week, double blind, placebo-controlled, randomized, multicenter, Phase 3 Clinical studies in CKD Stages 3 and 4 patients. Two studies used an identical three times a week dosing design, and one study used a daily dosing design. A total of 107 patients received Paricalcitol Capsules and 113 patients received placebo. The mean age of the patients was 63 years, 68% were male, 71% were Caucasian, and 26% were African-American. The average baseline iPTH was 274 pg/mL (range: 145-856 pg/mL). The average duration of CKD prior to study entry was 5.7 years. At study entry 22% were receiving calcium based phosphate binders and/or calcium supplements. Baseline 25-hydroxyvitamin D levels were not measured.
The initial dose of Paricalcitol Capsules was based on baseline iPTH. If iPTH was ≤ 500 pg/mL, Paricalcitol Capsules were administered 1 mcg daily or 2 mcg three times a week, not more than every other day. If iPTH was > 500 pg/mL, Paricalcitol Capsules were administered 2 mcg daily or 4 mcg three times a week, not more than every other day. The dose was increased by 1 mcg daily or 2 mcg three times a week every 2 to 4 weeks until iPTH levels were reduced by at least 30% from baseline. The overall average weekly dose of Paricalcitol Capsules was 9.6 mcg/week in the daily regimen and 9.5 mcg/week in the three times a week regimen.
In the clinical studies, doses were titrated for any of the following reasons: if iPTH fell to <60 pg/mL, or decreased > 60% from baseline, the dose was reduced or temporarily withheld; if iPTH decreased <30% from baseline and serum calcium was ≤10.3 mg/dL and serum phosphorus was ≤ 5.5 mg/dL, the dose was increased; and if iPTH decreased between 30 to 60% from baseline and serum calcium and phosphorus were ≤ 10.3 mg/dL and ≤ 5.5 mg/dL, respectively, the dose was maintained. Additionally, if serum calcium was between 10.4 to 11.0 mg/dL, the dose was reduced irrespective of iPTH, and the dose was withheld if serum calcium was > 11.0 mg/dL. If serum phosphorus was >5.5 mg/dL, dietary counseling was provided, and phosphate binders could have been initiated or increased. If the elevation persisted, the Paricalcitol Capsules dose was decreased. Seventy-seven percent (77%) of the Paricalcitol Capsules treated patients and 82% of the placebo treated patients completed the 24-week treatment. The primary efficacy endpoint of at least two consecutive ≥30% reductions from baseline iPTH was achieved by 91% of Paricalcitol Capsules treated patients and 13% of the placebo treated patients (p <0.001). The proportion of Paricalcitol Capsules treated patients achieving two consecutive ≥ 30% reductions was similar between the daily and the three times a week regimens (daily: 30/33, 91%; three times a week: 62/68, 91%).
The incidence of hypercalcemia (defined as two consecutive serum calcium values > 10.5 mg/dL), and hyperphosphatemia in Paricalcitol Capsules treated patients was similar to placebo. There were no treatment related adverse events associated with hypercalcemia or hyperphosphatemia in the Paricalcitol Capsules group. No increases in urinary calcium or phosphorous were detected in Paricalcitol Capsules treated patients compared to placebo.The pattern of change in the mean values for serum iPTH during the studies is shown in Figure 1
Figure 1. Mean Values for Serum iPTH Over Time in the Three Double-Blind, Placebo-Controlled, Phase 3, CKD Stages 3 and 4 Studies Combined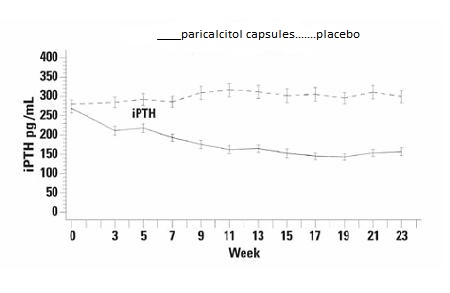
The mean changes from baseline to final treatment visit in serum iPTH, calcium, phosphorus, calcium-phosphorus product (Ca x P), and bone-specific alkaline phosphatase are shown in Table 7.
Table 7. Mean Changes from Baseline to Final Treatment Visit in Serum iPTH, Bone Specific Alkaline Phosphatase, Calcium, Phosphorus, and Calcium x Phosphorus Product In Three Combined Double-Blind, Placebo-Controlled, Phase 3, CKD Stages 3 and 4 Studies
Paricalcitol Capsules Placebo iPTH (pg/mL) n = 104 n = 110 Mean Baseline Value 266 279 Mean Change from Baseline (SE) -104 (9.2) +35 (9.0) Bone Specific Alkaline Phosphatase (mcg/L) n = 101 n = 107 Mean Baseline Value 17.1 18.8 Mean Final Treatment Value 9.2 17.4 Mean Change from Baseline (SE) -7.9 (0.76) -1.4(0.74) Calcium (mg/dL) n = 104 n = 110 Mean Baseline 9.3 9.4 Mean Final Treatment Value 9.5 9.3 Mean Change from Baseline (SE) +0.2 (0.04) -0.1(0.04) Phosphorous (mg/dL) n = 104 n = 110 Mean Baseline 4.0 4.0 Mean Final Treatment Value 4.3 4.3 Mean Change from Baseline (SE) +0.3 (0.08) +0.3 (0.08) Pediatric use information for patients 10 to 16 years of age is approved for AbbVie Inc.’s Zemplar (Paricalcitol) capsules. However, due to AbbVie Inc.’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
14.2 Chronic Kidney Disease Stage 5
AdultsThe safety and efficacy of Paricalcitol Capsules were evaluated in a Phase 3, 12-week, double blind, placebo-controlled, randomized, multicenter study in patients with CKD Stage 5 on HD or PD. The study used a three times a week dosing design. A total of 61 patients received Paricalcitol Capsules and 27 patients received placebo. The mean age of the patients was 57 years, 67% were male, 50% were Caucasian, 45% were African-American, and 53% were diabetic. The average baseline iPTH was 701 pg/mL (range: 216-1933 pg/mL). The average time since first dialysis across all subjects was 3.3 years.
The initial dose of Paricalcitol Capsules was based on baseline iPTH/60. Subsequent dose adjustments were based on iPTH/60 as well as primary chemistry results that were measured once a week. Starting at Treatment Week 2, study drug was maintained, increased or decreased weekly based on the results of the previous week’s calculation of iPTH/60. Paricalcitol Capsules were administered three times a week, not more than every other day.
The proportion of patients achieving at least two consecutive weekly ≥ 30% reductions from baseline iPTH was 88% of Paricalcitol Capsules treated patients and 13% of the placebo treated patients. The proportion of patients achieving at least two consecutive weekly ≥ 30% reductions from baseline iPTH was similar for HD and PD patients.
The incidence of hypercalcemia (defined as two consecutive serum calcium values > 10.5mg/dL) in patients treated with Paricalcitol Capsules was 6.6% as compared to 0% for patients given placebo. In PD patients the incidence of hypercalcemia in patients treated with Paricalcitol Capsules was 21% as compared to 0% for patients given placebo. The patterns of change in the mean values for serum iPTH are shown in Figure 2. The rate of hypercalcemia with Paricalcitol Capsules may be reduced with a lower dosing regimen based on the iPTH/80 formula as shown by computer simulations. The hypercalcemia rate can be further predicted to decrease, if the treatment is initiated in only those with baseline serum calcium ≤ 9.5 mg/dL [ see Clinical Pharmacology (12.2) and Dosage and Administration (2.2) ].
Figure 2. Mean Values for Serum iPTH Over Time in a Phase 3, Double-Blind, Placebo-Controlled CKD Stage 5 Study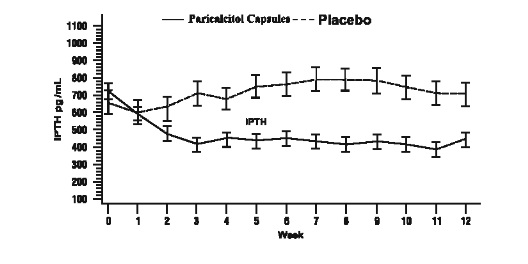
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Paricalcitol Capsules are available as 1 mcg, 2 mcg, and 4 mcg capsules.
The 1 mcg capsule is gray colour oval shaped soft gelatin capsule imprinted with ‘12’ in black ink and is available in the following package size.
Bottles of 30 (NDC 49483-687-03)
The 2 mcg capsule is brown colour oval shaped soft gelatin capsule imprinted with ‘14’ in black ink and is available in the following package size.
Bottles of 30 (NDC 49483-688-03)
The 4 mcg capsule is Light yellow colour oval shaped soft gelatin capsules imprinted with '17' in black ink and is available in the following package size.
Bottles of 30 (NDC 49483-699-03)
STORAGE
Store Paricalcitol Capsules at 20°C to 25°C (68°F to77°F). See USP Controlled Room Temperature.
-
17 PATIENT COUNSELING INFORMATION
Advise patients of the following:
· The most common adverse reactions with use of Paricalcitol Capsules, which include diarrhea, hypertension, nausea, nasopharyngitis dizziness and vomiting.
· Patients should adhere adhere to instructions regarding diet and phosphorus restriction.
· Patients should contact a health care provider if you develop symptoms of elevated calcium, (e.g. feeling tired, difficulty thinking clearly, loss of appetite, nausea, vomiting, constipation, increased thirst, increased urination and weight loss).
· Patients should return to the physician's office for routine monitoring. More frequent monitoring is necessary during the initiation of therapy, following dose changes or when potentially interacting medications are started or discontinued.
. Patients should inform their physician of all medications, including prescription and nonprescription drugs, supplements, and herbal preparations they are taking and any change to their medical condition. Patients should also inform their physician that they are taking Paricalcitol capsules if a new medication is prescribed.
· Breastfeeding is not recommended during treatment with Paricalcitol capsules [see Use in Specific Populations (8.2)].
Manufactured For:
Time-Cap Labs, Inc.
7 Michael Avenue, Farmingdale
New York 11735, USA
Made in India
Rev. 11/22 - PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
PARICALCITOL
paricalcitol capsule, liquid filledProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:49483-689 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PARICALCITOL (UNII: 6702D36OG5) (PARICALCITOL - UNII:6702D36OG5) PARICALCITOL 4 ug Inactive Ingredients Ingredient Name Strength MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) ALCOHOL (UNII: 3K9958V90M) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) GELATIN (UNII: 2G86QN327L) GLYCERIN (UNII: PDC6A3C0OX) SORBITOL (UNII: 506T60A25R) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) WATER (UNII: 059QF0KO0R) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) SHELLAC (UNII: MB5IUD6JUA) AMMONIA (UNII: 5138Q19F1X) Product Characteristics Color white (LIGHT YELLOW) Score no score Shape OVAL Size 10mm Flavor Imprint Code 17 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:49483-689-03 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA204948 01/03/2017 PARICALCITOL
paricalcitol capsule, liquid filledProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:49483-688 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PARICALCITOL (UNII: 6702D36OG5) (PARICALCITOL - UNII:6702D36OG5) PARICALCITOL 2 ug Inactive Ingredients Ingredient Name Strength MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) ALCOHOL (UNII: 3K9958V90M) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) GELATIN (UNII: 2G86QN327L) GLYCERIN (UNII: PDC6A3C0OX) SORBITOL (UNII: 506T60A25R) FERRIC OXIDE RED (UNII: 1K09F3G675) WATER (UNII: 059QF0KO0R) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) SHELLAC (UNII: MB5IUD6JUA) AMMONIA (UNII: 5138Q19F1X) Product Characteristics Color brown Score no score Shape OVAL Size 10mm Flavor Imprint Code 14 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:49483-688-03 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA204948 12/30/2015 PARICALCITOL
paricalcitol capsule, liquid filledProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:49483-687 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PARICALCITOL (UNII: 6702D36OG5) (PARICALCITOL - UNII:6702D36OG5) PARICALCITOL 1 ug Inactive Ingredients Ingredient Name Strength MEDIUM-CHAIN TRIGLYCERIDES (UNII: C9H2L21V7U) ALCOHOL (UNII: 3K9958V90M) BUTYLATED HYDROXYTOLUENE (UNII: 1P9D0Z171K) GELATIN (UNII: 2G86QN327L) GLYCERIN (UNII: PDC6A3C0OX) SORBITOL (UNII: 506T60A25R) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) WATER (UNII: 059QF0KO0R) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) FERROSOFERRIC OXIDE (UNII: XM0M87F357) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) SHELLAC (UNII: MB5IUD6JUA) AMMONIA (UNII: 5138Q19F1X) Product Characteristics Color gray Score no score Shape OVAL Size 9mm Flavor Imprint Code 12 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:49483-687-03 30 in 1 BOTTLE; Type 0: Not a Combination Product 01/03/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA204948 01/03/2017 Labeler - TIME CAP LABORATORIES, INC (037052099) Registrant - TIME CAP LABORATORIES, INC (037052099) Establishment Name Address ID/FEI Business Operations MARKSANS PHARMA LIMITED 925822975 manufacture(49483-689, 49483-688, 49483-687)


