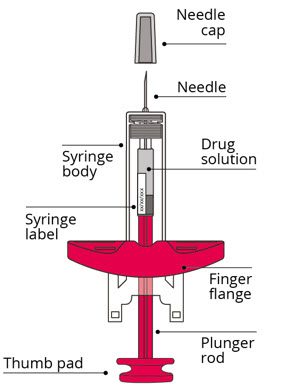
Label: AMVUTTRA- vutrisiran injection

- NDC Code(s): 71336-1003-1
- Packager: Alnylam Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated January 27, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use AMVUTTRA™ safely and effectively. See full prescribing information for AMVUTTRA.
AMVUTTRA (vutrisiran) injection, for subcutaneous use
Initial U.S. Approval: 2022INDICATIONS AND USAGE
AMVUTTRA is a transthyretin-directed small interfering RNA indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 25 mg/0.5 mL in a single-dose prefilled syringe. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
Reduced serum vitamin A levels and recommended supplementation: Supplement with the recommended daily allowance of vitamin A. Refer to an ophthalmologist if ocular symptoms suggestive of vitamin A deficiency occur. (5.1)
ADVERSE REACTIONS
The most common adverse reactions (≥5%) were pain in extremity, arthralgia, dyspnea, and vitamin A decreased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Alnylam Pharmaceuticals at 1-877-256-9526 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Reduced Serum Vitamin A Levels and Recommended Supplementation
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of AMVUTTRA is 25 mg administered by subcutaneous injection once every 3 months [see Dosage and Administration (2.2)].
2.2 Administration Instructions
AMVUTTRA is for subcutaneous use only and should be administered by a healthcare professional.
Preparation and Administration
- 1.
-
Prepare the syringe
If stored cold, allow the syringe to warm to room temperature for 30 minutes prior to use.
Remove the syringe from the packaging by gripping the syringe body.- Do not touch the plunger rod until ready to inject.
Check the following:- Syringe is not damaged, such as cracked or leaking
- Needle cap is attached to the syringe
- Expiration date on syringe label
- Do not use the syringe if any issues are found while checking the syringe.
- 2.
-
Choose and prepare the injection site
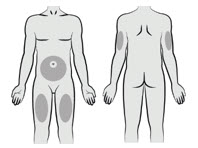
Choose an injection site from the following areas: the abdomen, thighs, or upper arms.
Avoid the following:- 5-cm area around the navel
- Scar tissue or areas that are reddened, inflamed, or swollen
- 3.
-
Prepare the syringe for injection
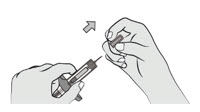
Hold the syringe body with one hand. Pull the needle cap straight off with other hand and dispose of needle cap immediately. It is normal to see a drop of liquid at the tip of the needle.- Do not touch the needle or let it touch any surface.
- Do not recap the syringe.
- Do not use the syringe if it is dropped.
- 4.
-
Perform the injection
Pinch the cleaned skin.
Fully insert the needle into the pinched skin at a 45°-90° angle.
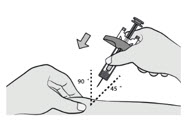
- Inject all of the medication.
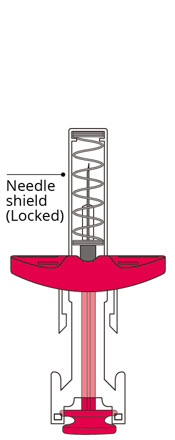
- Push the plunger rod as far as it will go to administer the dose and activate the needle shield.
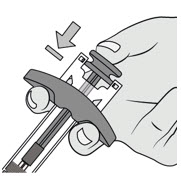
- Release the plunger rod to allow the needle shield to cover the needle.
- Do not block plunger rod movement.
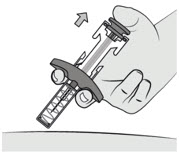
- 5.
-
Dispose of the syringe
Immediately dispose of the used syringe into a sharps container.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Reduced Serum Vitamin A Levels and Recommended Supplementation
AMVUTTRA treatment leads to a decrease in serum vitamin A levels [see Adverse Reactions (6.1) and Clinical Pharmacology (12.2)].
Supplementation at the recommended daily allowance of vitamin A is advised for patients taking AMVUTTRA. Higher doses than the recommended daily allowance of vitamin A should not be given to try to achieve normal serum vitamin A levels during treatment with AMVUTTRA, as serum vitamin A levels do not reflect the total vitamin A in the body.
Patients should be referred to an ophthalmologist if they develop ocular symptoms suggestive of vitamin A deficiency (e.g., night blindness).
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Reduced Serum Vitamin A Levels and Recommended Supplementation [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of AMVUTTRA cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In Study 1 [see Clinical Studies (14)], a total of 122 patients with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis (hATTR amyloidosis) received AMVUTTRA. Of these, 118 patients received at least 18 months of treatment. The mean duration of treatment was 18.8 months (range: 1.7 to 19.4 months). The median patient age at baseline was 60 years and 65% of the patients were male. Seventy percent of AMVUTTRA-treated patients were Caucasian, 17% were Asian, 3% were Black, and 9% were reported as Other. Forty-four percent of patients had the Val30Met mutation in the transthyretin gene; the remaining patients had one of 21 other mutations. At baseline, 70% of patients were in Stage 1 of the disease and 30% were in Stage 2.
The most common adverse reactions (at least 5%) were pain in extremity, arthralgia, dyspnea, and vitamin A decreased (see Table 1).
In Study 1, patients were instructed to take the recommended daily allowance of vitamin A [see Warnings and Precautions (5.1)]. Seventy-four percent of patients treated with AMVUTTRA had normal vitamin A levels at baseline, and 98% of those with a normal baseline developed low vitamin A levels. In some cases, the decreased vitamin A level was reported as an adverse reaction (see Table 1).
Table 1: Adverse Reactions Reported in at least 5% of Patients Treated with AMVUTTRA (Study 1) Adverse Reaction AMVUTTRA
N=122
%Pain in extremity* 15 Arthralgia* 11 Dyspnea* 7 Vitamin A decreased† 7 Two serious adverse reactions of atrioventricular (AV) heart block (1.6%) occurred in patients treated with AMVUTTRA, including one case of complete AV block.
Injection site reactions were reported in 5 (4%) patients treated with AMVUTTRA. Reported symptoms included bruising, erythema, pain, pruritus, and warmth. Injection site reactions were mild and transient.
6.2 Immunogenicity
As with all oligonucleotides, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In Study 1, 3 (2.5%) patients treated with AMVUTTRA developed anti-drug antibodies. Although anti-drug antibody development was not found to affect the pharmacokinetics, safety, or efficacy of AMVUTTRA in these patients, the available data are too limited to make definitive conclusions.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on AMVUTTRA use in pregnant women to inform a drug-associated risk of adverse developmental outcomes. AMVUTTRA treatment leads to a decrease in serum vitamin A levels, and vitamin A supplementation is advised for patients taking AMVUTTRA. Vitamin A is essential for normal embryofetal development; however, excessive levels of vitamin A are associated with adverse developmental effects. The effects on the fetus of a reduction in maternal serum TTR caused by AMVUTTRA and of vitamin A supplementation are unknown [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
In animal studies, subcutaneous administration of vutrisiran to pregnant rats resulted in developmental toxicity (reduced fetal body weight and embryofetal mortality) at doses associated with maternal toxicity (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Subcutaneous administration of vutrisiran (0, 3, 10, or 30 mg/kg/day) to pregnant rats during the period of organogenesis resulted in embryofetal mortality at the high dose and reduced fetal body weight at the mid and high doses, which were associated with maternal toxicity.
Subcutaneous administration of vutrisiran (0, 3, 10, or 30 mg/kg/day) to pregnant rabbits resulted in no adverse effects on embryofetal development.
Subcutaneous administration of vutrisiran (0, 5, 10, or 20 mg/kg) to pregnant rats every 6 days throughout pregnancy and lactation resulted in no adverse developmental effects on the offspring.
8.2 Lactation
Risk Summary
There is no information regarding the presence of vutrisiran in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for AMVUTTRA and any potential adverse effects on the breastfed infant from AMVUTTRA or from the underlying maternal condition.
8.5 Geriatric Use
No dose adjustment is required in patients ≥65 years of age [see Clinical Pharmacology (12.3)]. A total of 46 (38%) patients ≥65 years of age, including 7 (6%) patients ≥75 years of age, received AMVUTTRA in Study 1. No overall differences in safety or effectiveness were observed between these patients and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dose adjustment is recommended in patients with mild or moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥30 to <90 mL/min/1.73 m2) [see Clinical Pharmacology (12.3)]. AMVUTTRA has not been studied in patients with severe renal impairment or end-stage renal disease.
8.7 Hepatic Impairment
No dose adjustment is recommended in patients with mild hepatic impairment (total bilirubin ≤1 × ULN and AST >1 × ULN, or total bilirubin >1.0 to 1.5 × ULN and any AST) [see Clinical Pharmacology (12.3)]. AMVUTTRA has not been studied in patients with moderate or severe hepatic impairment.
-
11 DESCRIPTION
AMVUTTRA contains vutrisiran, a chemically modified double-stranded small interfering ribonucleic acid (siRNA) that targets mutant and wild-type transthyretin (TTR) messenger RNA (mRNA) and is covalently linked to a ligand containing three N-acetylgalactosamine (GalNAc) residues to enable delivery of the siRNA to hepatocytes. The structural formula of vutrisiran sodium is presented below.
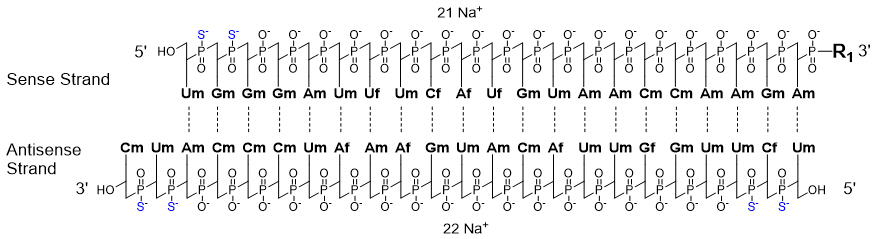
Af, Cf, Gf, Uf = 2'-F ribonucleosides
Am, Cm, Gm, Um = 2'-OMe ribonucleosides
Dashed lines denote Watson-Crick base pairingvutrisiran 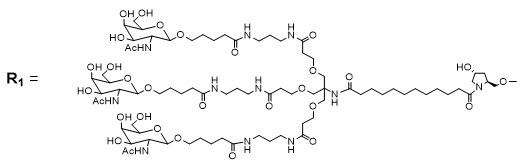
The molecular formula of vutrisiran sodium is C530H672F9N171Na43O323P43S6 with a molecular weight of 17,290 Da. The molecular formula of the free acid is C530H715F9N171O323P43S6 with a molecular weight of 16,345 Da.
AMVUTTRA is supplied as a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous injection. Each 0.5 mL of solution contains 25 mg of vutrisiran (equivalent to 26.5 mg vutrisiran sodium), 0.2 mg sodium phosphate monobasic dihydrate, 0.7 mg sodium phosphate dibasic dihydrate, 3.2 mg sodium chloride, water for injection, and sodium hydroxide and/or phosphoric acid to adjust the pH to ~7.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Vutrisiran is a double-stranded siRNA-GalNAc conjugate that causes degradation of mutant and wild-type TTR mRNA through RNA interference, which results in a reduction of serum TTR protein and TTR protein deposits in tissues.
12.2 Pharmacodynamics
In Study 1 [see Clinical Studies (14)], following administration of the recommended AMVUTTRA dosage every 3 months to patients with hATTR amyloidosis, vutrisiran reduced mean serum TTR at steady state by 83%. Similar TTR reductions were observed regardless of Val30Met genotype status, weight, sex, age, or race.
Vutrisiran also reduced the mean steady state serum vitamin A by 62% over 9 months [see Warnings and Precautions (5.1)].
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of AMVUTTRA were evaluated following a single dose in healthy subjects and multiple doses in patients with hATTR amyloidosis, as summarized in Table 2.
Table 2: Pharmacokinetic Parameters of Vutrisiran Vutrisiran AUCinf = area under the concentration-time curve from the time of dosing extrapolated to infinity; AUClast = area under the concentration-time curve from the time of dosing to the last measurable concentration; Cmax = maximum plasma concentration; CV = coefficient of variation; RSE = relative standard error; Tmax = time to maximum concentration; Vd/F = apparent volume of distribution - *
- After 25 mg every 3 months dosage in hATTR amyloidosis patients
- †
- After 25 mg single dose in healthy subjects
- ‡
- Based on population PK model estimation
- §
- Vutrisiran plasma protein binding was concentration-dependent and decreased with increasing vutrisiran concentrations (from 78% at 0.5 mcg/mL to 19% at 50 mcg/mL)
- ¶
- After single subcutaneous vutrisiran dose from 5 to 300 mg (i.e., 0.2 to 12 times the recommended dose) in healthy subjects
General Information Dose Proportionality Vutrisiran Cmax showed dose proportional increase while AUClast and AUCinf were slightly more than dose proportional following single subcutaneous doses ranging from 5 to 300 mg (i.e., 0.2 to 12 times the recommended dose) Accumulation No accumulation of vutrisiran was observed in plasma after repeated every 3 months dosage* Absorption Tmax [Median (Range)] 4 (0.17, 12.0) hours† Distribution Estimated Vd/F (%RSE) 10.1 (5.8) L‡ Protein Binding 80%§ Organ Distribution Vutrisiran distributes primarily to the liver after subcutaneous dosing Elimination Half-Life [Median (Range)] 5.2 (2.2, 6.4) hours† Apparent Clearance [Median (Range)] 21.4 (19.8, 30) L/hour† Metabolism Primary Pathway Vutrisiran is metabolized by endo- and exonucleases to short nucleotide fragments of varying sizes within the liver Excretion Primary Pathway The mean fraction of unchanged vutrisiran eliminated in urine was approximately 19.4% at the recommended dose of 25 mg. The mean renal clearance of vutrisiran ranged from 4.5 to 5.7 L/hour¶ Specific Populations
No clinically significant differences in the pharmacokinetics of vutrisiran were observed based on age, sex, race, mild and moderate renal impairment (eGFR≥30 to <90 mL/min/1.73 m2), or mild hepatic impairment (total bilirubin ≤1 × ULN and AST >1 × ULN, or total bilirubin >1.0 to 1.5 × ULN and any AST). Vutrisiran has not been studied in patients with severe renal impairment, end-stage renal disease, moderate or severe hepatic impairment, or in patients with prior liver transplant.
Drug Interaction Studies
No clinical drug-drug interaction studies have been performed with vutrisiran. In vitro studies suggest that vutrisiran is not a substrate or inhibitor of cytochrome P450 enzymes. Vutrisiran is not expected to cause drug-drug interactions by inducing CYP enzymes or modulating the activities of drug transporters.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Subcutaneous administration of vutrisiran to male rats (0, 4, 7.5, or 15 mg/kg once every 4 weeks or 15 mg/kg once every 12 weeks) for 99 weeks and to female rats (0, 6, 12.5, or 25 mg/kg once every 4 weeks or 25 mg/kg once every 12 weeks) for 86-87 weeks resulted in no increase in tumors.
-
14 CLINICAL STUDIES
The efficacy of AMVUTTRA was evaluated in a randomized, open-label clinical trial in adult patients with polyneuropathy caused by hATTR amyloidosis (Study 1; NCT03759379). Patients were randomized 3:1 to receive 25 mg of AMVUTTRA subcutaneously once every 3 months (N=122), or 0.3 mg/kg patisiran intravenously every 3 weeks (N=42) as a reference group. Ninety-seven percent of AMVUTTRA-treated patients and 93% of patisiran-treated patients completed at least 9 months of the assigned treatment.
Efficacy assessments were based on a comparison of the AMVUTTRA arm of Study 1 with an external placebo group in another study (NCT01960348) composed of a comparable population of adult patients with polyneuropathy caused by hATTR amyloidosis.
The primary efficacy endpoint was the change from baseline to Month 9 in modified Neuropathy Impairment Score +7 (mNIS+7). The mNIS+7 is an objective assessment of neuropathy and comprises the NIS and Modified +7 composite scores. In the version of the mNIS+7 used in the trial, the NIS objectively measures deficits in cranial nerve function, muscle strength, and reflexes, and the +7 assesses postural blood pressure, quantitative sensory testing, and peripheral nerve electrophysiology. The mNIS+7 has a total score range from 0 to 304 points, with higher scores representing a greater severity of disease.
The clinical meaningfulness of effects on the mNIS+7 was assessed by the change from baseline to Month 9 in Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) total score. The Norfolk QoL-DN scale is a patient-reported assessment that evaluates the subjective experience of neuropathy in the following domains: physical functioning/large fiber neuropathy, activities of daily living, symptoms, small fiber neuropathy, and autonomic neuropathy. The Norfolk QoL-DN has a total score range from -4 to 136, with higher scores representing greater impairment.
Additional endpoints were gait speed, as measured by the 10-meter walk test (10MWT), and modified body mass index (mBMI).
Treatment with AMVUTTRA in Study 1 resulted in statistically significant improvements in the mNIS+7, Norfolk QoL-DN total score, and 10-meter walk test at Month 9 compared to placebo in the external study (p<0.001) [Table 3, Figure 1, and Figure 3]. The distributions of changes in mNIS+7 and Norfolk QoL-DN total scores from baseline to Month 9 by percent of patients are shown in Figure 2 and Figure 4, respectively.
The change from baseline to Month 9 in modified body mass index nominally favored AMVUTTRA [Table 3].
Table 3: Clinical Efficacy Results (Comparison of AMVUTTRA Treatment in Study 1 to an External Placebo Control*) Endpoint† Baseline, Mean (SD) Change from Baseline to Month 9, LS Mean (SEM) AMVUTTRA-Placebo* Treatment Difference, LS Mean
(95% CI)p-value AMVUTTRA
N=122
(Study 1)Placebo*
N=77
(NCT01960348)AMVUTTRA
(Study 1)Placebo*
(NCT01960348)CI = confidence interval; LS mean = least squares mean; mBMI = modified body mass index; mNIS = modified Neuropathy Impairment Score; QoL-DN = Quality of Life-Diabetic Neuropathy; SD = standard deviation; SEM = standard error of the mean - *
- External placebo group from another randomized controlled trial (NCT01960348)
- †
- All endpoints analyzed using the analysis of covariance (ANCOVA) with multiple imputation (MI) method)
- ‡
- A lower number indicates less impairment/fewer symptoms
- §
- A higher number indicates less disability/less impairment
- ¶
- mBMI: nominal p-value; body mass index (BMI; kg/m2) multiplied by serum albumin (g/L).
mNIS+7‡ 60.6 (36.0) 74.6 (37.0) -2.2 (1.4) 14.8 (2.0) -17.0
(-21.8, -12.2)p<0.001 Norfolk
QoL-DN‡47.1 (26.3) 55.5 (24.3) -3.3 (1.7) 12.9 (2.2) -16.2
(-21.7, -10.8)p<0.001 10-meter walk test (m/sec)§ 1.01 (0.39) 0.79 (0.32) 0 (0.02) -0.13 (0.03) 0.13
(0.07, 0.19)p<0.001 mBMI¶ 1058 (234) 990 (214) 7.6 (7.9) -60.2 (10.1) 67.8
(43.0, 92.6)p<0.001 Figure 1: Change from Baseline in mNIS+7 (Comparison of AMVUTTRA Treatment in Study 1 to an External Placebo Control*) A decrease in mNIS+7 indicates improvement
Δ indicates between-group treatment difference, shown as the LS mean difference (95% CI) for AMVUTTRA – placebo- *
- External placebo group from another randomized controlled trial (NCT01960348)
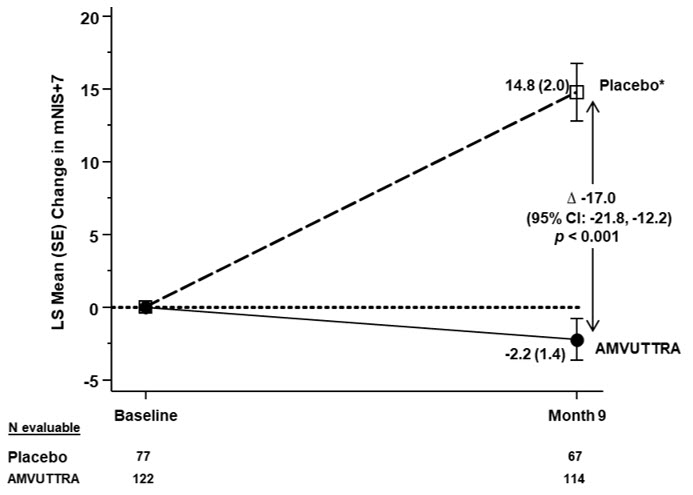
Figure 2: Histogram of mNIS+7 Change from Baseline at Month 9 (Comparison of AMVUTTRA Treatment in Study 1 to an External Placebo Control*) Categories are mutually exclusive; patients who died before 9 months are summarized in the "Death" category only - *
- External placebo group from another randomized controlled trial (NCT01960348)
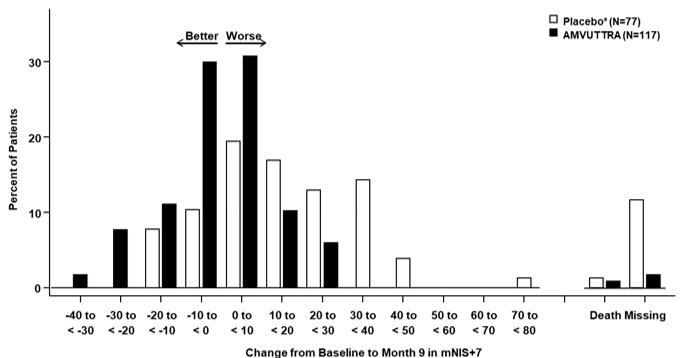
Figure 3: Change from Baseline in Norfolk QoL-DN Total Score (Comparison of AMVUTTRA Treatment in Study 1 to an External Placebo Control*) A decrease in Norfolk QoL-DN score indicates improvement
Δ indicates between-group treatment difference, shown as the LS mean difference (95% CI) for AMVUTTRA – placebo- *
- External placebo group from another randomized controlled trial (NCT01960348)
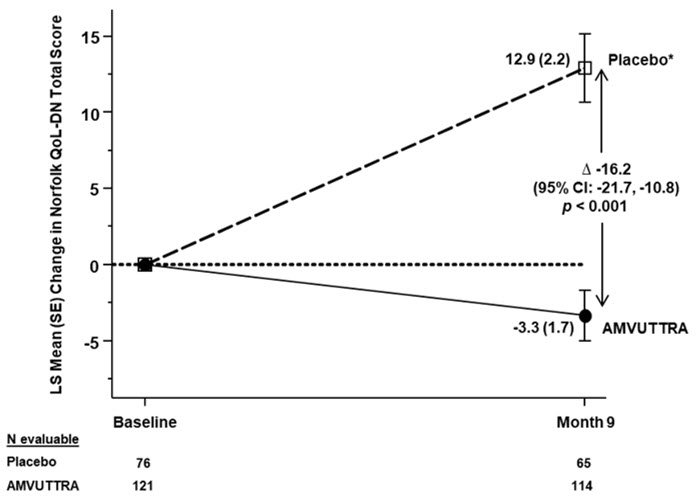
Figure 4: Histogram of Norfolk QoL-DN Total Score Change from Baseline at Month 9 (Comparison of AMVUTTRA Treatment in Study 1 to an External Placebo Control*) Categories are mutually exclusive; patients who died before 9 months are summarized in the "Death" category only - *
- External placebo group from another randomized controlled trial (NCT01960348)
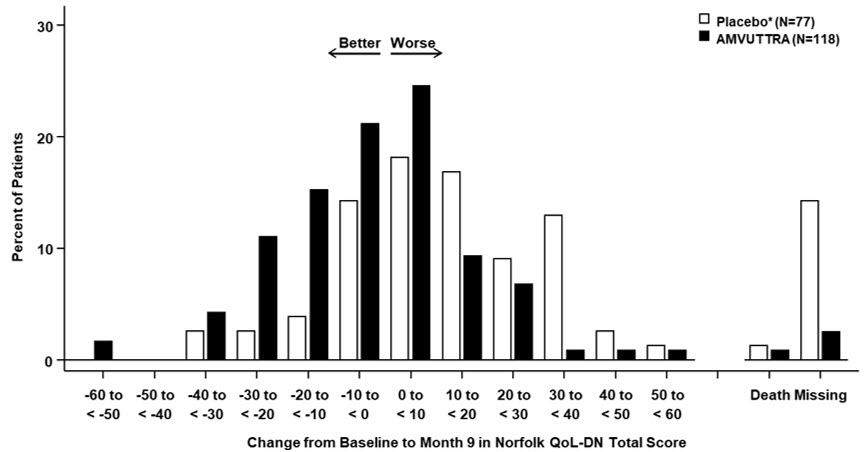
Patients receiving AMVUTTRA in Study 1 experienced similar improvements relative to those in the external placebo group in mNIS+7 and Norfolk QoL-DN total score across all subgroups including age, sex, race, region, NIS score, Val30Met genotype status, and disease stage.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
AMVUTTRA is a sterile, preservative-free, clear, colorless-to-yellow solution for subcutaneous injection. AMVUTTRA is supplied as 25 mg/0.5 mL solution in a single-dose 1-mL prefilled syringe made from Type I glass with stainless steel 29-gauge needle with a needle shield. The prefilled syringe components are not made with natural rubber latex.
AMVUTTRA is available in cartons containing one single-dose prefilled syringe each.
The NDC is: 71336-1003-1.
-
17 PATIENT COUNSELING INFORMATION
Recommended Vitamin A Supplementation
Inform patients that AMVUTTRA treatment leads to a decrease in serum vitamin A levels. Instruct patients to take the recommended daily allowance of vitamin A. Advise patients to contact their healthcare provider if they experience ocular symptoms suggestive of vitamin A deficiency (e.g., night blindness) and refer them to an ophthalmologist if they develop these symptoms [see Warnings and Precautions (5.1)].
Pregnancy
Instruct patients that if they are pregnant or plan to become pregnant while taking AMVUTTRA they should inform their healthcare provider. Inform patients of the potential risk to the fetus, including that AMVUTTRA treatment leads to a decrease in serum vitamin A levels [see Use in Specific Populations (8.1) and Clinical Pharmacology (12.2)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 25 mg Syringe Carton
-
INGREDIENTS AND APPEARANCE
AMVUTTRA
vutrisiran injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71336-1003 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VUTRISIRAN (UNII: GB4I2JI8UI) (VUTRISIRAN - UNII:GB4I2JI8UI) VUTRISIRAN 25 mg in 0.5 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Sodium hydroxide (UNII: 55X04QC32I) Phosphoric acid (UNII: E4GA8884NN) SODIUM PHOSPHATE, MONOBASIC, DIHYDRATE (UNII: 5QWK665956) SODIUM PHOSPHATE, DIBASIC, DIHYDRATE (UNII: 94255I6E2T) Sodium chloride (UNII: 451W47IQ8X) Product Characteristics Color YELLOW (clear, colorless-to-yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71336-1003-1 1 in 1 CARTON 06/13/2022 1 0.5 mL in 1 SYRINGE, GLASS; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215515 06/13/2022 Labeler - Alnylam Pharmaceuticals, Inc. (115524410)