Label: PROCRIT- erythropoietin injection, solution







-
NDC Code(s):
59676-302-00,
59676-302-01,
59676-303-00,
59676-303-01, view more59676-304-00, 59676-304-01, 59676-310-00, 59676-310-01, 59676-310-02, 59676-312-00, 59676-312-04, 59676-320-00, 59676-320-04, 59676-340-00, 59676-340-01
- Packager: Janssen Products, LP
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated May 9, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PROCRIT safely and effectively. See full prescribing information for PROCRIT.
PROCRIT ®(epoetin alfa) injection, for intravenous or subcutaneous use
Initial U.S. Approval: 1989WARNING: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE
See full prescribing information for complete boxed warning.
Chronic Kidney Disease:
- In controlled trials, patients experienced greater risks for death, serious adverse cardiovascular reactions, and stroke when administered erythropoiesis-stimulating agents (ESAs) to target a hemoglobin level of greater than 11 g/dL ( 5.1).
- No trial has identified a hemoglobin target level, ESA dose, or dosing strategy that does not increase these risks ( 2.2).
- Use the lowest PROCRIT dose sufficient to reduce the need for red blood cell (RBC) transfusions ( 5.1).
Cancer:
- ESAs shortened overall survival and/or increased the risk of tumor progression or recurrence in clinical studies of patients with breast, non-small cell lung, head and neck, lymphoid, and cervical cancers ( 5.2).
- Use the lowest dose to avoid RBC transfusions ( 2.4).
- Use ESAs only for anemia from myelosuppressive chemotherapy ( 1.3).
- ESAs are not indicated for patients receiving myelosuppressive chemotherapy when the anticipated outcome is cure ( 1.5).
- Discontinue following the completion of a chemotherapy course ( 2.4).
Perisurgery:
- Due to increased risk of deep venous thrombosis (DVT), DVT prophylaxis is recommended ( 5.1).
INDICATIONS AND USAGE
PROCRIT is an erythropoiesis-stimulating agent (ESA) indicated for:
- Treatment of anemia due to
- Chronic Kidney Disease (CKD) in patients on dialysis and not on dialysis ( 1.1).
- Zidovudine in patients with Human Immunodeficiency Virus (HIV) infection ( 1.2).
- The effects of concomitant myelosuppressive chemotherapy, and upon initiation, there is a minimum of two additional months of planned chemotherapy ( 1.3).
- Reduction of allogeneic red blood cell (RBC) transfusions in patients undergoing elective, noncardiac, nonvascular surgery ( 1.4).
Limitations of Use
PROCRIT has not been shown to improve quality of life, fatigue, or patient well-being ( 1.5).
PROCRIT is not indicated for use:
- In patients with cancer receiving hormonal agents, biologic products, or radiotherapy, unless also receiving concomitant myelosuppressive chemotherapy ( 1.5).
- In patients with cancer receiving myelosuppressive chemotherapy when the anticipated outcome is cure ( 1.5).
- In patients with cancer receiving myelosuppressive chemotherapy in whom the anemia can be managed by transfusion ( 1.5).
- In patients scheduled for surgery who are willing to donate autologous blood ( 1.5).
- In patients undergoing cardiac or vascular surgery ( 1.5).
- As a substitute for RBC transfusions in patients who require immediate correction of anemia ( 1.5).
DOSAGE AND ADMINISTRATION
- Evaluate iron status before and during treatment and maintain iron repletion. Correct or exclude other causes of anemia before initiating treatment ( 2.1).
- In pregnant women, lactating women, neonates, infants: Use only single-dose vials ( 2.1).
- Patients with CKD: Initial dose: 50 to 100 Units/kg 3 times weekly (adults) and 50 Units/kg 3 times weekly (pediatric patients). Individualize maintenance dose. Intravenous route recommended for patients on hemodialysis ( 2.2).
- Patients on Zidovudine due to HIV Infection: 100 Units/kg 3 times weekly ( 2.3).
- Patients with Cancer on Chemotherapy: 40,000 Units weekly or 150 Units/kg 3 times weekly (adults); 600 Units/kg intravenously weekly (pediatric patients ≥ 5 years) ( 2.4).
- Surgery Patients: 300 Units/kg per day daily for 15 days or 600 Units/kg weekly ( 2.5).
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- Uncontrolled hypertension ( 4)
- Pure red cell aplasia (PRCA) that begins after treatment with PROCRIT or other erythropoietin protein drugs ( 4)
- Serious allergic reactions to PROCRIT ( 4)
- Use of the multiple-dose vials containing benzyl alcohol in neonates, infants, pregnant women, and lactating women ( 4)
WARNINGS AND PRECAUTIONS
- Increased Mortality, Myocardial Infarction, Stroke, and Thromboembolism: Using ESAs to target a hemoglobin level of greater than 11 g/dL increases the risk of serious adverse cardiovascular reactions and has not been shown to provide additional benefit ( 5.1and 14.1). Use caution in patients with coexistent cardiovascular disease and stroke ( 5.1).
- Increased Mortality and/or Increased Risk of Tumor Progression or Recurrence in Patients with Cancer ( 5.2).
- Hypertension: Control hypertension prior to initiating and during treatment with PROCRIT ( 5.3).
- Seizures: PROCRIT increases the risk for seizures in patients with CKD ( 5.4). Increase monitoring of these patients for changes in seizure frequency or premonitory symptoms ( 5.4).
- PRCA: If severe anemia and low reticulocyte count develop during PROCRIT treatment, withhold PROCRIT and evaluate for PRCA ( 5.6).
- Serious Allergic Reactions: Discontinue PROCRIT and manage reactions ( 5.7).
- Severe Cutaneous Reactions: Discontinue PROCRIT ( 5.8).
ADVERSE REACTIONS
- Patients with CKD: Adverse reactions in ≥ 5% of PROCRIT-treated patients in clinical studies were hypertension, arthralgia, muscle spasm, pyrexia, dizziness, medical device malfunction, vascular occlusion, and upper respiratory tract infection ( 6.1).
- Patients on Zidovudine due to HIV Infection: Adverse reactions in ≥ 5% of PROCRIT-treated patients in clinical studies were pyrexia, cough, rash, and injection site irritation ( 6.1).
- Patients with Cancer on Chemotherapy: Adverse reactions in ≥ 5% of PROCRIT-treated patients in clinical studies were nausea, vomiting, myalgia, arthralgia, stomatitis, cough, weight decrease, leukopenia, bone pain, rash, hyperglycemia, insomnia, headache, depression, dysphagia, hypokalemia, and thrombosis ( 6.1).
- Surgery Patients: Adverse reactions in ≥ 5% of PROCRIT-treated patients in clinical studies were nausea, vomiting, pruritus, headache, injection site pain, chills, deep vein thrombosis, cough, and hypertension ( 6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Products, LP at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE
1 INDICATIONS AND USAGE
1.1 Anemia Due to Chronic Kidney Disease
1.2 Anemia Due to Zidovudine in Patients with HIV Infection
1.3 Anemia Due to Chemotherapy in Patients with Cancer
1.4 Reduction of Allogeneic Red Blood Cell Transfusions in Patients Undergoing Elective, Noncardiac, Nonvascular Surgery
1.5 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Patients with Chronic Kidney Disease
2.3 Zidovudine-treated Patients with HIV Infection
2.4 Patients on Cancer Chemotherapy
2.5 Surgery Patients
2.6 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Increased Mortality, Myocardial Infarction, Stroke, and Thromboembolism
5.2 Increased Mortality and/or Increased Risk of Tumor Progression or Recurrence in Patients with Cancer
5.3 Hypertension
5.4 Seizures
5.5 Lack or Loss of Hemoglobin Response to PROCRIT
5.6 Pure Red Cell Aplasia
5.7 Serious Allergic Reactions
5.8 Severe Cutaneous Reactions
5.9 Risk of Serious Adverse Reactions Due to Benzyl Alcohol Preservative
5.10 Risk of Infectious Diseases Due to Albumin (Human) Content
5.11 Dialysis Management
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
6.3 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Patients with Chronic Kidney Disease
14.2 Zidovudine-treated Patients with HIV Infection
14.3 Patients with Cancer on Chemotherapy
14.4 Surgery Patients
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: ESAs INCREASE THE RISK OF DEATH, MYOCARDIAL INFARCTION, STROKE, VENOUS THROMBOEMBOLISM, THROMBOSIS OF VASCULAR ACCESS AND TUMOR PROGRESSION OR RECURRENCE
Chronic Kidney Disease:
- In controlled trials, patients with chronic kidney disease (CKD) experienced greater risks for death, serious adverse cardiovascular reactions, and stroke when administered erythropoiesis-stimulating agents (ESAs) to target a hemoglobin level of greater than 11 g/dL[see Warnings and Precautions (5.1)] .
- No trial has identified a hemoglobin target level, ESA dose, or dosing strategy that does not increase these risks[see Dosage and Administration (2.2)] .
- Use the lowest PROCRIT dose sufficient to reduce the need for red blood cell (RBC) transfusions[see Warnings and Precautions (5.1)] .
Cancer:
- ESAs shortened overall survival and/or increased the risk of tumor progression or recurrence in clinical studies of patients with breast, non-small cell lung, head and neck, lymphoid, and cervical cancers[see Warnings and Precautions (5.2)] .
- To decrease these risks, as well as the risk of serious cardiovascular and thromboembolic reactions, use the lowest dose needed to avoid RBC transfusions [see Dosage and Administration (2.4)] .
- Use ESAs only for anemia from myelosuppressive chemotherapy[see Indications and Usage (1.3)] .
- ESAs are not indicated for patients receiving myelosuppressive chemotherapy when the anticipated outcome is cure[see Indications and Usage (1.5)] .
- Discontinue following the completion of a chemotherapy course[see Dosage and Administration (2.4)] .
Perisurgery:
- Due to increased risk of Deep Venous Thrombosis (DVT), DVT prophylaxis is recommended[see Dosage and Administration (2.5), Warnings and Precautions (5.1)] .
-
1 INDICATIONS AND USAGE
1.1 Anemia Due to Chronic Kidney Disease
PROCRIT is indicated for the treatment of anemia due to chronic kidney disease (CKD), including patients on dialysis and not on dialysis to decrease the need for red blood cell (RBC) transfusion.
1.2 Anemia Due to Zidovudine in Patients with HIV Infection
PROCRIT is indicated for the treatment of anemia due to zidovudine administered at ≤ 4200 mg/week in patients with HIV Infection with endogenous serum erythropoietin levels of ≤ 500 mUnits/mL.
1.3 Anemia Due to Chemotherapy in Patients with Cancer
PROCRIT is indicated for the treatment of anemia in patients with non-myeloid malignancies where anemia is due to the effect of concomitant myelosuppressive chemotherapy, and upon initiation, there is a minimum of two additional months of planned chemotherapy.
1.4 Reduction of Allogeneic Red Blood Cell Transfusions in Patients Undergoing Elective, Noncardiac, Nonvascular Surgery
PROCRIT is indicated to reduce the need for allogeneic RBC transfusions among patients with perioperative hemoglobin > 10 to ≤ 13 g/dL who are at high risk for perioperative blood loss from elective, noncardiac, nonvascular surgery. PROCRIT is not indicated for patients who are willing to donate autologous blood pre-operatively.
1.5 Limitations of Use
PROCRIT has not been shown to improve quality of life, fatigue, or patient well-being.
PROCRIT is not indicated for use:
- In patients with cancer receiving hormonal agents, biologic products, or radiotherapy, unless also receiving concomitant myelosuppressive chemotherapy.
- In patients with cancer receiving myelosuppressive chemotherapy when the anticipated outcome is cure.
- In patients with cancer receiving myelosuppressive chemotherapy in whom the anemia can be managed by transfusion.
- In patients scheduled for surgery who are willing to donate autologous blood.
- In patients undergoing cardiac or vascular surgery.
- As a substitute for RBC transfusions in patients who require immediate correction of anemia.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
Evaluation of Iron Stores and Nutritional Factors
Evaluate the iron status in all patients before and during treatment. Administer supplemental iron therapy when serum ferritin is less than 100 mcg/L or when serum transferrin saturation is less than 20%. The majority of patients with CKD will require supplemental iron during the course of ESA therapy.
Monitoring of Response to Therapy
Correct or exclude other causes of anemia (e.g., vitamin deficiency, metabolic or chronic inflammatory conditions, bleeding, etc.) before initiating PROCRIT. Following initiation of therapy and after each dose adjustment, monitor hemoglobin weekly until the hemoglobin level is stable and sufficient to minimize the need for RBC transfusion.
Selection of Formulation
In pregnant women, lactating women, neonates, and infants use only single-dose vials (the benzyl alcohol-free formulation) [see Contraindications (4)and Use in Specific Populations (8.1, 8.2, and 8.4)].
2.2 Patients with Chronic Kidney Disease
In controlled trials, patients experienced greater risks for death, serious adverse cardiovascular reactions, and stroke when administered ESAs to target a hemoglobin level of greater than 11 g/dL. No trial has identified a hemoglobin target level, ESA dose, or dosing strategy that does not increase these risks. Individualize dosing and use the lowest dose of PROCRIT sufficient to reduce the need for RBC transfusions [see Warnings and Precautions (5.1)] . Physicians and patients should weigh the possible benefits of decreasing transfusions against the increased risks of death and other serious cardiovascular adverse reactions [see Boxed Warningand Clinical Studies (14)].
For all patients with CKD:
When initiating or adjusting therapy, monitor hemoglobin levels at least weekly until stable, then monitor at least monthly. When adjusting therapy consider hemoglobin rate of rise, rate of decline, ESA responsiveness and hemoglobin variability. A single hemoglobin excursion may not require a dosing change.
- Do not increase the dose more frequently than once every 4 weeks. Decreases in dose can occur more frequently. Avoid frequent dose adjustments.
- If the hemoglobin rises rapidly (e.g., more than 1 g/dL in any 2-week period), reduce the dose of PROCRIT by 25% or more as needed to reduce rapid responses.
- For patients who do not respond adequately, if the hemoglobin has not increased by more than 1 g/dL after 4 weeks of therapy, increase the dose by 25%.
- For patients who do not respond adequately over a 12-week escalation period, increasing the PROCRIT dose further is unlikely to improve response and may increase risks. Use the lowest dose that will maintain a hemoglobin level sufficient to reduce the need for RBC transfusions. Evaluate other causes of anemia. Discontinue PROCRIT if responsiveness does not improve.
For adult patients with CKD on dialysis:
- Initiate PROCRIT treatment when the hemoglobin level is less than 10 g/dL.
- If the hemoglobin level approaches or exceeds 11 g/dL, reduce or interrupt the dose of PROCRIT.
- The recommended starting dose for adult patients is 50 to 100 Units/kg 3 times weekly intravenously or subcutaneously. The intravenous route is recommended for patients on hemodialysis.
For adult patients with CKD not on dialysis:
- Consider initiating PROCRIT treatment only when the hemoglobin level is less than 10 g/dL
andthe following considerations apply:
- The rate of hemoglobin decline indicates the likelihood of requiring a RBC transfusion and,
- Reducing the risk of alloimmunization and/or other RBC transfusion-related risks is a goal
- If the hemoglobin level exceeds 10 g/dL, reduce or interrupt the dose of PROCRIT, and use the lowest dose of PROCRIT sufficient to reduce the need for RBC transfusions.
- The recommended starting dose for adult patients is 50 to 100 Units/kg 3 times weekly intravenously or subcutaneously.
For pediatric patients with CKD:
- Initiate PROCRIT treatment only when the hemoglobin level is less than 10 g/dL.
- If the hemoglobin level approaches or exceeds 12 g/dL, reduce or interrupt the dose of PROCRIT.
- The recommended starting dose for pediatric patients (ages 1 month or older) is 50 Units/kg 3 times weekly intravenously or subcutaneously.
When treating patients who have chronic kidney disease and cancer, physicians should refer to Warnings and Precautions (5.1 and 5.2).
2.3 Zidovudine-treated Patients with HIV Infection
Starting Dose
The recommended starting dose in adults is 100 Units/kg as an intravenous or subcutaneous injection 3 times per week.
Dose Adjustment
- If hemoglobin does not increase after 8 weeks of therapy, increase PROCRIT dose by approximately 50 to 100 Units/kg at 4- to 8-week intervals until hemoglobin reaches a level needed to avoid RBC transfusions or 300 Units/kg.
- Withhold PROCRIT if hemoglobin exceeds 12 g/dL. Resume therapy at a dose 25% below the previous dose when hemoglobin declines to less than 11 g/dL.
Discontinue PROCRIT if an increase in hemoglobin is not achieved at a dose of 300 Units/kg for 8 weeks.
2.4 Patients on Cancer Chemotherapy
Initiate PROCRIT in patients on cancer chemotherapy only if the hemoglobin is less than 10 g/dL, and if there is a minimum of two additional months of planned chemotherapy.
Use the lowest dose of PROCRIT necessary to avoid RBC transfusions.
Recommended Starting Dose
Dose Reduction
Reduce dose by 25% if:
- Hemoglobin increases greater than 1 g/dL in any 2-week period or
- Hemoglobin reaches a level needed to avoid RBC transfusion.
Withhold dose if hemoglobin exceeds a level needed to avoid RBC transfusion. Reinitiate at a dose 25% below the previous dose when hemoglobin approaches a level where RBC transfusions may be required.
Dose Increase
After the initial 4 weeks of PROCRIT therapy, if hemoglobin increases by less than 1 g/dL andremains below 10 g/dL, increase dose to:
- 300 Units/kg three times per week in adults or
- 60,000 Units weekly in adults
- 900 Units/kg (maximum 60,000 Units) weekly in pediatric patients
After 8 weeks of therapy, if there is no response as measured by hemoglobin levels or if RBC transfusions are still required, discontinue PROCRIT.
2.5 Surgery Patients
The recommended PROCRIT regimens are:
- 300 Units/kg per day subcutaneously for 15 days total: administered daily for 10 days before surgery, on the day of surgery, and for 4 days after surgery.
- 600 Units/kg subcutaneously in 4 doses administered 21, 14, and 7 days before surgery and on the day of surgery.
Deep venous thrombosis prophylaxis is recommended during PROCRIT therapy [see Warnings and Precautions (5.1)] .
2.6 Preparation and Administration
- Do not shake. Do not use PROCRIT that has been shaken or frozen.
- Protect vials from light.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Do not use any vials exhibiting particulate matter or discoloration.
- Discard unused portions of PROCRIT in preservative-free vials. Do not re-enter preservative-free vials.
- Store unused portions of PROCRIT in multiple-dose vials at 36°F to 46°F (2°C to 8°C). Discard 21 days after initial entry.
- Do not dilute. Do not mix with other drug solutions except for admixing as described below:
Preservative-free PROCRIT from single-dose vials may be admixed in a syringe with bacteriostatic 0.9% sodium chloride injection, USP, with benzyl alcohol 0.9% (bacteriostatic saline) in a 1:1 ratio using aseptic technique at the time of administration. Do not mix PROCRIT with bacteriostatic saline when administering to pregnant women, lactating women, neonates, and infants [see Use in Specific Populations (8.1, 8.2, 8.4)].
-
3 DOSAGE FORMS AND STRENGTHS
Injection:
- 2,000 Units/mL, 3,000 Units/mL, 4,000 Units/mL, 10,000 Units/mL, and 40,000 Units/mL of PROCRIT as a clear and colorless liquid in single-dose vials
- 20,000 Units/2 mL (10,000 Units/mL) and 20,000 Units/mL of PROCRIT as a clear and colorless liquid in multiple-dose vials (contains benzyl alcohol)
-
4 CONTRAINDICATIONS
PROCRIT is contraindicated in patients with:
- Uncontrolled hypertension [see Warnings and Precautions (5.3)]
- Pure red cell aplasia (PRCA) that begins after treatment with PROCRIT or other erythropoietin protein drugs [see Warnings and Precautions (5.6)]
- Serious allergic reactions to PROCRIT [see Warnings and Precautions (5.7)]
PROCRIT from multiple-dose vials contains benzyl alcohol and is contraindicated in:
- Neonates, infants, pregnant women, and lactating women [see Warnings and Precautions (5.9), Use in Specific Populations (8.1, 8.2, 8.4)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Increased Mortality, Myocardial Infarction, Stroke, and Thromboembolism
- In controlled clinical trials of patients with CKD comparing higher hemoglobin targets (13 – 14 g/dL) to lower targets (9 – 11.3 g/dL), PROCRIT and other ESAs increased the risk of death, myocardial infarction, stroke, congestive heart failure, thrombosis of hemodialysis vascular access, and other thromboembolic events in the higher target groups.
- Using ESAs to target a hemoglobin level of greater than 11 g/dL increases the risk of serious adverse cardiovascular reactions and has not been shown to provide additional benefit [see Clinical Studies (14.1)] . Use caution in patients with coexistent cardiovascular disease and stroke [see Dosage and Administration (2.2)] . Patients with CKD and an insufficient hemoglobin response to ESA therapy may be at even greater risk for cardiovascular reactions and mortality than other patients. A rate of hemoglobin rise of greater than 1 g/dL over 2 weeks may contribute to these risks.
- In controlled clinical trials of patients with cancer, PROCRIT and other ESAs increased the risks for death and serious adverse cardiovascular reactions. These adverse reactions included myocardial infarction and stroke.
- In controlled clinical trials, ESAs increased the risk of death in patients undergoing coronary artery bypass graft surgery (CABG) and the risk of deep venous thrombosis (DVT) in patients undergoing orthopedic procedures.
The design and overall results of the 3 large trials comparing higher and lower hemoglobin targets are shown in Table 1.
Table 1: Randomized Controlled Trials Showing Adverse Cardiovascular Outcomes in Patients With CKD Normal Hematocrit Study (NHS)
(N=1265)CHOIR
(N=1432)TREAT
(N=4038)Time Period of Trial 1993 to 1996 2003 to 2006 2004 to 2009 Population CKD patients on hemodialysis with coexisting CHF or CAD, hematocrit 30 ± 3% on epoetin alfa CKD patients not on dialysis with hemoglobin < 11 g/dL not previously administered epoetin alfa CKD patients not on dialysis with type II diabetes, hemoglobin ≤ 11 g/dL Hemoglobin Target;
Higher vs. Lower (g/dL)14.0 vs. 10.0 13.5 vs. 11.3 13.0 vs. ≥ 9.0 Median (Q1, Q3)
Achieved Hemoglobin level (g/dL)12.6 (11.6, 13.3) vs. 10.3 (10.0, 10.7) 13.0 (12.2, 13.4) vs. 11.4 (11.1, 11.6) 12.5 (12.0, 12.8) vs. 10.6 (9.9, 11.3) Primary Endpoint All-cause mortality or non-fatal MI All-cause mortality, MI, hospitalization for CHF, or stroke All-cause mortality, MI, myocardial ischemia, heart failure, and stroke Hazard Ratio or Relative Risk (95% CI) 1.28 (1.06 – 1.56) 1.34 (1.03 – 1.74) 1.05 (0.94 – 1.17) Adverse Outcome for Higher Target Group All-cause mortality All-cause mortality Stroke Hazard Ratio or Relative Risk (95% CI) 1.27 (1.04 – 1.54) 1.48 (0.97 – 2.27) 1.92 (1.38 – 2.68) Patients with Chronic Kidney Disease
Normal Hematocrit Study (NHS): A prospective, randomized, open-label study of 1265 patients with chronic kidney disease on dialysis with documented evidence of congestive heart failure or ischemic heart disease was designed to test the hypothesis that a higher target hematocrit (Hct) would result in improved outcomes compared with a lower target Hct. In this study, patients were randomized to epoetin alfa treatment targeted to a maintenance hemoglobin of either 14 ± 1 g/dL or 10 ± 1 g/dL. The trial was terminated early with adverse safety findings of higher mortality in the high hematocrit target group. Higher mortality (35% vs. 29%) was observed for the patients randomized to a target hemoglobin of 14 g/dL than for the patients randomized to a target hemoglobin of 10 g/dL. For all-cause mortality, the HR=1.27; 95% CI (1.04, 1.54); p=0.018. The incidence of nonfatal myocardial infarction, vascular access thrombosis, and other thrombotic events was also higher in the group randomized to a target hemoglobin of 14 g/dL.
CHOIR: A randomized, prospective trial, 1432 patients with anemia due to CKD who were not undergoing dialysis and who had not previously received epoetin alfa therapy were randomized to epoetin alfa treatment targeting a maintenance hemoglobin concentration of either 13.5 g/dL or 11.3 g/dL. The trial was terminated early with adverse safety findings. A major cardiovascular event (death, myocardial infarction, stroke, or hospitalization for congestive heart failure) occurred in 125 of the 715 patients (18%) in the higher hemoglobin group compared to 97 of the 717 patients (14%) in the lower hemoglobin group [hazard ratio (HR) 1.34, 95% CI: 1.03, 1.74; p=0.03].
TREAT: A randomized, double-blind, placebo-controlled, prospective trial of 4038 patients with: CKD not on dialysis (eGFR of 20 – 60 mL/min), anemia (hemoglobin levels ≤ 11 g/dL), and type 2 diabetes mellitus, patients were randomized to receive either darbepoetin alfa treatment or a matching placebo. Placebo group patients also received darbepoetin alfa when their hemoglobin levels were below 9 g/dL. The trial objectives were to demonstrate the benefit of darbepoetin alfa treatment of the anemia to a target hemoglobin level of 13 g/dL, when compared to a "placebo" group, by reducing the occurrence of either of two primary endpoints: (1) a composite cardiovascular endpoint of all-cause mortality or a specified cardiovascular event (myocardial ischemia, CHF, MI, and CVA) or (2) a composite renal endpoint of all-cause mortality or progression to end stage renal disease. The overall risks for each of the two primary endpoints (the cardiovascular composite and the renal composite) were not reduced with darbepoetin alfa treatment (see Table 1), but the risk of stroke was increased nearly two-fold in the darbepoetin alfa -treated group versus the placebo group: annualized stroke rate 2.1% vs. 1.1%, respectively, HR 1.92; 95% CI: 1.38, 2.68; p < 0.001. The relative risk of stroke was particularly high in patients with a prior stroke: annualized stroke rate 5.2% in the darbepoetin alfa-treated group and 1.9% in the placebo group, HR 3.07; 95% CI: 1.44, 6.54. Also, among darbepoetin alfa-treated subjects with a past history of cancer, there were more deaths due to all causes and more deaths adjudicated as due to cancer, in comparison with the control group.
Patients with Cancer
An increased incidence of thromboembolic reactions, some serious and life-threatening, occurred in patients with cancer treated with ESAs.
In a randomized, placebo-controlled study (Study 2 in Table 2 [see Warnings and Precautions (5.2)] ) of 939 women with metastatic breast cancer receiving chemotherapy, patients received either weekly epoetin alfa or placebo for up to a year. This study was designed to show that survival was superior when epoetin alfa was administered to prevent anemia (maintain hemoglobin levels between 12 and 14 g/dL or hematocrit between 36% and 42%). This study was terminated prematurely when interim results demonstrated a higher mortality at 4 months (8.7% vs. 3.4%) and a higher rate of fatal thrombotic reactions (1.1% vs. 0.2%) in the first 4 months of the study among patients treated with epoetin alfa. Based on Kaplan-Meier estimates, at the time of study termination, the 12-month survival was lower in the epoetin alfa group than in the placebo group (70% vs. 76%; HR 1.37, 95% CI: 1.07, 1.75; p=0.012).
Patients Having Surgery
An increased incidence of deep venous thrombosis (DVT) in patients receiving epoetin alfa undergoing surgical orthopedic procedures was demonstrated [see Adverse Reactions (6.1)] . In a randomized, controlled study, 680 adult patients, not receiving prophylactic anticoagulation and undergoing spinal surgery, were randomized to 4 doses of 600 Units/kg epoetin alfa (7, 14, and 21 days before surgery, and the day of surgery) and standard of care (SOC) treatment (n=340) or to SOC treatment alone (n=340). A higher incidence of DVTs, determined by either color flow duplex imaging or by clinical symptoms, was observed in the epoetin alfa group (16 [4.7%] patients) compared with the SOC group (7 [2.1%] patients). In addition to the 23 patients with DVTs included in the primary analysis, 19 [2.8%] patients (n=680) experienced 1 other thrombovascular event (TVE) each (12 [3.5%] in the epoetin alfa group and 7 [2.1%] in the SOC group). Deep venous thrombosis prophylaxis is strongly recommended when ESAs are used for the reduction of allogeneic RBC transfusions in surgical patients [see Dosage and Administration (2.5)] .
Increased mortality was observed in a randomized, placebo-controlled study of PROCRIT in adult patients who were undergoing CABG surgery (7 deaths in 126 patients randomized to PROCRIT versus no deaths among 56 patients receiving placebo). Four of these deaths occurred during the period of study drug administration and all 4 deaths were associated with thrombotic events.
5.2 Increased Mortality and/or Increased Risk of Tumor Progression or Recurrence in Patients with Cancer
ESAs resulted in decreased locoregional control/progression-free survival (PFS) and/or overall survival (OS) (see Table 2).
Adverse effects on PFS and/or OS were observed in studies of patients receiving chemotherapy for breast cancer (Studies 1, 2, and 4), lymphoid malignancy (Study 3), and cervical cancer (Study 5); in patients with advanced head and neck cancer receiving radiation therapy (Studies 6 and 7); and in patients with non-small cell lung cancer or various malignancies who were not receiving chemotherapy or radiotherapy (Studies 8 and 9).
Table 2: Randomized, Controlled Studies with Decreased Survival and/or Decreased Locoregional Control Study/Tumor/(n) Hemoglobin Target Achieved Hemoglobin
(Median; Q1, Q3 *)Primary Efficacy Outcome Adverse Outcome for ESA- containing Arm Chemotherapy Study 1
Metastatic breast cancer
(n=2098)≤12 g/dL † 11.6 g/dL
10.7, 12.1 g/dLProgression-free survival (PFS) Decreased progression-free and overall survival Study 2
Metastatic breast cancer
(n=939)12–14 g/dL 12.9 g/dL;
12.2, 13.3 g/dL12-month overall survival Decreased 12-month survival Study 3
Lymphoid malignancy
(n=344)13–15 g/dL (M)
13–14 g/dL (F)11 g/dL;
9.8, 12.1 g/dLProportion of patients achieving a hemoglobin response Decreased overall survival Study 4
Early breast cancer
(n=733)12.5–13 g/dL 13.1 g/dL;
12.5, 13.7 g/dLRelapse-free and overall survival Decreased 3-year relapse-free and overall survival Study 5
Cervical cancer
(n=114)12–14 g/dL 12.7 g/dL;
12.1, 13.3 g/dLProgression-free and overall survival and locoregional control Decreased 3-year progression-free and overall survival and locoregional control Radiotherapy Alone Study 6
Head and neck cancer
(n=351)≥ 15 g/dL (M)
≥ 14 g/dL (F)Not available Locoregional progression-free survival Decreased 5-year locoregional progression-free and overall survival Study 7
Head and neck cancer
(n=522)14–15.5 g/dL Not available Locoregional disease control Decreased locoregional disease control No Chemotherapy or Radiotherapy Study 8
Non-small cell lung cancer
(n=70)12–14 g/dL Not available Quality of life Decreased overall survival Study 9
Non-myeloid malignancy
(n=989)12–13 g/dL 10.6 g/dL;
9.4, 11.8 g/dLRBC transfusions Decreased overall survival Decreased Overall Survival
Study 2 was described in the previous section [see Warnings and Precautions (5.1)] . Mortality at 4 months (8.7% vs. 3.4%) was significantly higher in the epoetin alfa arm. The most common investigator-attributed cause of death within the first 4 months was disease progression; 28 of 41 deaths in the epoetin alfa arm and 13 of 16 deaths in the placebo arm were attributed to disease progression. Investigator-assessed time to tumor progression was not different between the 2 groups. Survival at 12 months was significantly lower in the epoetin alfa arm (70% vs. 76%; HR 1.37, 95% CI: 1.07, 1.75; p=0.012).
Study 3 was a randomized, double-blind study (darbepoetin alfa vs. placebo) conducted in 344 anemic patients with lymphoid malignancy receiving chemotherapy. With a median follow-up of 29 months, overall mortality rates were significantly higher among patients randomized to darbepoetin alfa as compared to placebo (HR 1.36, 95% CI: 1.02, 1.82).
Study 8 was a multicenter, randomized, double-blind study (epoetin alfa vs. placebo) in which patients with advanced non-small cell lung cancer receiving only palliative radiotherapy or no active therapy were treated with epoetin alfa to achieve and maintain hemoglobin levels between 12 and 14 g/dL. Following an interim analysis of 70 patients (planned accrual 300 patients), a significant difference in survival in favor of the patients in the placebo arm of the study was observed (median survival 63 vs. 129 days; HR 1.84; p=0.04).
Study 9 was a randomized, double-blind study (darbepoetin alfa vs. placebo) in 989 anemic patients with active malignant disease, neither receiving nor planning to receive chemotherapy or radiation therapy. There was no evidence of a statistically significant reduction in proportion of patients receiving RBC transfusions. The median survival was shorter in the darbepoetin alfa treatment group than in the placebo group (8 months vs. 10.8 months; HR 1.30, 95% CI: 1.07, 1.57).
Decreased Progression-free Survival and Overall Survival
Study 1 was a randomized, open-label, multicenter study in 2,098 anemic women with metastatic breast cancer, who received first line or second line chemotherapy. This was a noninferiority study designed to rule out a 15% risk increase in tumor progression or death of epoetin alfa plus standard of care (SOC) as compared with SOC alone. At the time of clinical data cutoff, the median progression free survival (PFS) per investigator assessment of disease progression was 7.4 months in each arm (HR 1.09, 95% CI: 0.99, 1.20), indicating the study objective was not met. There were more deaths from disease progression in the epoetin alfa plus SOC arm (59% vs. 56%) and more thrombotic vascular events in the epoetin alfa plus SOC arm (3% vs. 1%). At the final analysis, 1653 deaths were reported (79.8% subjects in the epoetin alfa plus SOC group and 77.8% subjects in the SOC group). Median overall survival in the epoetin alfa plus SOC group was 17.8 months compared with 18.0 months in the SOC alone group (HR 1.07, 95% CI: 0.97, 1.18).
Study 4 was a randomized, open-label, controlled, factorial design study in which darbepoetin alfa was administered to prevent anemia in 733 women receiving neo-adjuvant breast cancer treatment. A final analysis was performed after a median follow-up of approximately 3 years. The 3-year survival rate was lower (86% vs. 90%; HR 1.42, 95% CI: 0.93, 2.18) and the 3-year relapse-free survival rate was lower (72% vs. 78%; HR 1.33, 95% CI: 0.99, 1.79) in the darbepoetin alfa-treated arm compared to the control arm.
Study 5 was a randomized, open-label, controlled study that enrolled 114 of a planned 460 cervical cancer patients receiving chemotherapy and radiotherapy. Patients were randomized to receive epoetin alfa to maintain hemoglobin between 12 and 14 g/dL or to RBC transfusion support as needed. The study was terminated prematurely due to an increase in thromboembolic adverse reactions in epoetin alfa-treated patients compared to control (19% vs. 9%). Both local recurrence (21% vs. 20%) and distant recurrence (12% vs. 7%) were more frequent in epoetin alfa-treated patients compared to control. Progression-free survival at 3 years was lower in the epoetin alfa-treated group compared to control (59% vs. 62%; HR 1.06, 95% CI: 0.58, 1.91). Overall survival at 3 years was lower in the epoetin alfa-treated group compared to control (61% vs. 71%; HR 1.28, 95% CI: 0.68, 2.42).
Study 6 was a randomized, placebo-controlled study in 351 head and neck cancer patients where epoetin beta or placebo was administered to achieve target hemoglobins ≥ 14 and ≥ 15 g/dL for women and men, respectively. Locoregional progression-free survival was significantly shorter in patients receiving epoetin beta (HR 1.62, 95% CI: 1.22, 2.14; p=0.0008) with medians of 406 days and 745 days in the epoetin beta and placebo arms, respectively. Overall survival was significantly shorter in patients receiving epoetin beta (HR 1.39, 95% CI: 1.05, 1.84; p=0.02).
Decreased Locoregional Control
Study 7 was a randomized, open-label, controlled study conducted in 522 patients with primary squamous cell carcinoma of the head and neck receiving radiation therapy alone (no chemotherapy) who were randomized to receive darbepoetin alfa to maintain hemoglobin levels of 14 to 15.5 g/dL or no darbepoetin alfa. An interim analysis performed on 484 patients demonstrated that locoregional control at 5 years was significantly shorter in patients receiving darbepoetin alfa (RR 1.44, 95% CI: 1.06, 1.96; p=0.02). Overall survival was shorter in patients receiving darbepoetin alfa (RR 1.28, 95% CI: 0.98, 1.68; p=0.08).
5.3 Hypertension
PROCRIT is contraindicated in patients with uncontrolled hypertension. Following initiation and titration of PROCRIT, approximately 25% of patients on dialysis required initiation of or increases in antihypertensive therapy; hypertensive encephalopathy and seizures have been reported in patients with CKD receiving PROCRIT.
Appropriately control hypertension prior to initiation of and during treatment with PROCRIT. Reduce or withhold PROCRIT if blood pressure becomes difficult to control. Advise patients of the importance of compliance with antihypertensive therapy and dietary restrictions [see Patient Counseling Information (17)] .
5.4 Seizures
PROCRIT increases the risk of seizures in patients with CKD. During the first several months following initiation of PROCRIT, monitor patients closely for premonitory neurologic symptoms. Advise patients to contact their healthcare practitioner for new-onset seizures, premonitory symptoms or change in seizure frequency.
5.5 Lack or Loss of Hemoglobin Response to PROCRIT
For lack or loss of hemoglobin response to PROCRIT, initiate a search for causative factors (e.g., iron deficiency, infection, inflammation, bleeding). If typical causes of lack or loss of hemoglobin response are excluded, evaluate for PRCA [see Warnings and Precautions (5.6)] . In the absence of PRCA, follow dosing recommendations for management of patients with an insufficient hemoglobin response to PROCRIT therapy [see Dosage and Administration (2.2)] .
5.6 Pure Red Cell Aplasia
Cases of PRCA and of severe anemia, with or without other cytopenias that arise following the development of neutralizing antibodies to erythropoietin have been reported in patients treated with PROCRIT. This has been reported predominantly in patients with CKD receiving ESAs by subcutaneous administration. PRCA has also been reported in patients receiving ESAs for anemia related to hepatitis C treatment (an indication for which PROCRIT is not approved).
If severe anemia and low reticulocyte count develop during treatment with PROCRIT, withhold PROCRIT and evaluate patients for neutralizing antibodies to erythropoietin. Contact Janssen Products, LP at 1-800-JANSSEN (1-800-526-7736) to perform assays for binding and neutralizing antibodies. Permanently discontinue PROCRIT in patients who develop PRCA following treatment with PROCRIT or other erythropoietin protein drugs. Do not switch patients to other ESAs.
5.7 Serious Allergic Reactions
Serious allergic reactions, including anaphylactic reactions, angioedema, bronchospasm, skin rash, and urticaria may occur with PROCRIT. Immediately and permanently discontinue PROCRIT and administer appropriate therapy if a serious allergic or anaphylactic reaction occurs.
5.8 Severe Cutaneous Reactions
Blistering and skin exfoliation reactions including Erythema multiforme and Stevens-Johnson Syndrome (SJS)/Toxic Epidermal Necrolysis (TEN), have been reported in patients treated with ESAs (including PROCRIT) in the postmarketing setting. Discontinue PROCRIT therapy immediately if a severe cutaneous reaction, such as SJS/TEN, is suspected.
5.9 Risk of Serious Adverse Reactions Due to Benzyl Alcohol Preservative
PROCRIT from multiple-dose vials contains benzyl alcohol and is contraindicated for use in neonates, infants, pregnant women, and lactating women [see Contraindications (4)] . In addition, do not mix PROCRIT with bacteriostatic saline (which also contains benzyl alcohol) when administering PROCRIT to these patient populations [see Dosage and Administration (2)].
Serious and fatal reactions including "gasping syndrome" can occur in neonates and infants treated with benzyl alcohol-preserved drugs, including PROCRIT multiple-dose vials. The "gasping syndrome" is characterized by central nervous system (CNS) depression, metabolic acidosis, and gasping respirations. There is a potential for similar risks to fetuses and infants exposed to benzyl alcohol in uteroor in breast-fed milk, respectively. PROCRIT multiple-dose vials contain 11 mg of benzyl alcohol per mL. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Use in Specific Populations (8.1, 8.2, and 8.4)] .
5.10 Risk of Infectious Diseases Due to Albumin (Human) Content
PROCRIT contains albumin, a derivative of human blood [see Description (11)] . Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases. A theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD) also is considered extremely remote. No cases of transmission of viral diseases or CJD have ever been identified for albumin.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Increased Mortality, Myocardial Infarction, Stroke, and Thromboembolism [see Warnings and Precautions (5.1)]
- Increased Mortality and/or Increased Risk of Tumor Progression or Recurrence in Patients with Cancer [see Warnings and Precautions (5.2)]
- Hypertension [see Warnings and Precautions (5.3)]
- Seizures [see Warnings and Precautions (5.4)]
- PRCA [see Warnings and Precautions (5.6)]
- Serious Allergic Reactions [see Warnings and Precautions (5.7)]
- Severe Cutaneous Reactions [see Warnings and Precautions (5.8)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of other drugs and may not reflect the rates observed in practice.
Patients with Chronic Kidney Disease
Adult Patients
Three double-blind, placebo-controlled studies, including 244 patients with CKD on dialysis, were used to identify the adverse reactions to PROCRIT. In these studies, the mean age of patients was 48 years (range: 20 to 80 years). One hundred and thirty-three (55%) patients were men. The racial distribution was as follows: 177 (73%) patients were white, 48 (20%) patients were black, 4 (2%) patients were Asian, 12 (5%) patients were other, and racial information was missing for 3 (1%) patients.
Two double-blind, placebo-controlled studies, including 210 patients with CKD not on dialysis, were used to identify the adverse reactions to PROCRIT. In these studies, the mean age of patients was 57 years (range: 24 to 79 years). One hundred and twenty-one (58%) patients were men. The racial distribution was as follows: 164 (78%) patients were white, 38 (18%) patients were black, 3 (1%) patients were Asian, 3 (1%) patients were other, and racial information was missing for 2 (1%) patients.
The adverse reactions with a reported incidence of ≥ 5% in PROCRIT-treated patients and that occurred at a ≥ 1% higher frequency than in placebo-treated patients are shown in the table below:
Table 3: Adverse Reactions in Patients with CKD on Dialysis Adverse Reaction PROCRIT-treated Patients
(n=148)Placebo-treated Patients
(n=96)Hypertension 27.7% 12.5% Arthralgia 16.2% 3.1% Muscle spasm 7.4% 6.3% Pyrexia 10.1% 8.3% Dizziness 9.5% 8.3% Medical Device Malfunction (artificial kidney clotting during dialysis) 8.1% 4.2% Vascular Occlusion (vascular access thrombosis) 8.1% 2.1% Upper respiratory tract infection 6.8% 5.2% An additional serious adverse reaction that occurred in less than 5% of epoetin alfa-treated dialysis patients and greater than placebo was thrombosis (2.7% PROCRIT and 1% placebo) [see Warnings and Precautions (5.1)].
The adverse reactions with a reported incidence of ≥ 5% in PROCRIT-treated patients and that occurred at a ≥ 1% higher frequency than in placebo-treated patients are shown in the table below:
Table 4: Adverse Reactions in Patients with CKD Not on Dialysis Adverse Reactions PROCRIT-treated Patients
(n=131)Placebo-treated Patients
(n=79)Hypertension 13.7% 10.1% Arthralgia 12.2% 7.6% Additional serious adverse reactions that occurred in less than 5% of epoetin alfa-treated patients not on dialysis and greater than placebo were erythema (0.8% PROCRIT and 0% placebo) and myocardial infarction (0.8% PROCRIT and 0% placebo) [see Warnings and Precautions (5.1)].
Zidovudine-treated Patients with HIV Infection
A total of 297 zidovudine-treated patients with HIV Infection were studied in 4 placebo-controlled studies. A total of 144 (48%) patients were randomly assigned to receive PROCRIT and 153 (52%) patients were randomly assigned to receive placebo. PROCRIT was administered at doses between 100 and 200 Units/kg 3 times weekly subcutaneously for up to 12 weeks.
For the combined PROCRIT treatment groups, a total of 141 (98%) men and 3 (2%) women between the ages of 24 and 64 years were enrolled. The racial distribution of the combined PROCRIT treatment groups was as follows: 129 (90%) white, 8 (6%) black, 1 (1%) Asian, and 6 (4%) other.
In double-blind, placebo-controlled studies of 3 months duration involving approximately 300 zidovudine-treated patients with HIV Infection, adverse reactions with an incidence of ≥ 1% in patients treated with PROCRIT were:
Table 5: Adverse Reactions in Zidovudine-treated Patients with HIV Infection Adverse Reaction PROCRIT
(n=144)Placebo
(n=153)Pyrexia 42% 34% Cough 26% 14% Rash 19% 7% Injection site irritation 7% 4% Urticaria 3% 1% Respiratory tract congestion 1% Not reported Pulmonary embolism 1% Not reported Patients with Cancer on Chemotherapy
The data below were obtained in Study C1, a 16-week, double-blind, placebo-controlled study that enrolled 344 patients with anemia secondary to chemotherapy. There were 333 patients who were evaluable for safety; 168 of 174 patients (97%) randomized to PROCRIT received at least 1 dose of study drug, and 165 of 170 patients (97%) randomized to placebo received at least 1 placebo dose. For the once weekly PROCRIT-treatment group, a total of 76 men (45%) and 92 women (55%) between the ages of 20 and 88 years were treated. The racial distribution of the PROCRIT-treatment group was 158 white (94%) and 10 black (6%). PROCRIT was administered once weekly for an average of 13 weeks at a dose of 20,000 to 60,000 IU subcutaneously (mean weekly dose was 49,000 IU).
The adverse reactions with a reported incidence of ≥ 5% in PROCRIT-treated patients that occurred at a higher frequency than in placebo-treated patients are shown in the table below:
Table 6: Adverse Reactions in Patients with Cancer Adverse Reaction PROCRIT
(n=168)Placebo
(n=165)Nausea 35% 30% Vomiting 20% 16% Myalgia 10% 5% Arthralgia 10% 6% Stomatitis 10% 8% Cough 9% 7% Weight decrease 9% 5% Leukopenia 8% 7% Bone pain 7% 4% Rash 7% 5% Hyperglycemia 6% 4% Insomnia 6% 2% Headache 5% 4% Depression 5% 4% Dysphagia 5% 2% Hypokalemia 5% 3% Thrombosis 5% 3% Surgery Patients
Four hundred sixty-one patients undergoing major orthopedic surgery were studied in a placebo-controlled study (S1) and a comparative dosing study (2 dosing regimens, S2). A total of 358 patients were randomly assigned to receive PROCRIT and 103 (22%) patients were randomly assigned to receive placebo. PROCRIT was administered daily at a dose of 100 to 300 IU/kg subcutaneously for 15 days or at 600 IU/kg once weekly for 4 weeks.
For the combined PROCRIT treatment groups, a total of 90 (25%) men and 268 (75%) women between the ages of 29 and 89 years were enrolled. The racial distribution of the combined PROCRIT treatment groups was as follows: 288 (80%) white, 64 (18%) black, 1 (< 1%) Asian, and 5 (1%) other.
The adverse reactions with a reported incidence of ≥ 1% in PROCRIT-treated patients that occurred at a higher frequency than in placebo-treated patients are shown in the table below:
Table 7: Adverse Reactions in Surgery Patients Adverse Reaction Study S1 Study S2 PROCRIT
300 U/kgPROCRIT
100 U/kgPlacebo PROCRIT
600 U/kg × 4 weeksPROCRIT
300 U/kg × 15 days(n=112) * (n=101) * (n=103) * (n=73) † (n=72) † Nausea 47% 43% 45% 45% 56% Vomiting 21% 12% 14% 19% 28% Pruritus 16% 16% 14% 12% 21% Headache 13% 11% 9% 10% 18% Injection site pain 13% 9% 8% 12% 11% Chills 7% 4% 1% 1% 0% Deep vein thrombosis 6% 3% 3% 0% ‡ 0% ‡ Cough 5% 4% 0% 4% 4% Hypertension 5% 3% 5% 5% 6% Rash 2% 2% 1% 3% 3% Edema 1% 2% 2% 1% 3% 6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of PROCRIT.
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Seizures [see Warnings and Precautions (5.4)]
- PRCA [see Warnings and Precautions (5.6)]
- Serious Allergic Reactions [see Warnings and Precautions (5.7)]
- Injection Site Reactions, including irritation and pain
- Porphyria
- Severe Cutaneous Reactions [see Warnings and Precautions (5.8)]
6.3 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to epoetin alfa with the incidence of antibodies to other products may be misleading.
Neutralizing antibodies to epoetin alfa that cross-react with endogenous erythropoietin and other ESAs can result in PRCA or severe anemia (with or without other cytopenias) [see Warnings and Precautions (5.6)] .
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
PROCRIT from multiple-dose vials contains benzyl alcohol and is contraindicated in pregnant women [see Contraindications (4)] . When therapy with PROCRIT is needed during pregnancy, use a benzyl alcohol-free formulation (i.e., single-dose vial). Do not mix PROCRIT with bacteriostatic saline when administering to pregnant women because it contains benzyl alcohol (see Clinical Considerations) [see Dosage and Administration (2.1)] .
The limited available data on PROCRIT use in pregnant women are insufficient to determine a drug-associated risk of adverse developmental outcomes. In animal reproductive and developmental toxicity studies, adverse fetal effects including embryo-fetal death, skeletal anomalies, and growth defects occurred when pregnant rats received epoetin alfa at doses approximating the clinical recommended starting doses (see Data) . Consider the benefits and risks of PROCRIT single-dose vials for the mother and possible risks to the fetus when prescribing PROCRIT to a pregnant woman.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. . All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
The multiple-dose vials of PROCRIT contain benzyl alcohol. The preservative benzyl alcohol has been associated with serious adverse reactions and death when administered intravenously to neonates and infants [see Warnings and Precautions (5.9), Use in Specific Populations (8.4)]. There is a potential for similar risks to fetuses exposed to benzyl alcohol in utero.
Data
Human Data
There are reports of pregnant women with anemia alone or anemia associated with severe renal disease and other hematologic disorders who received PROCRIT. Polyhydramnios and intrauterine growth restriction were reported in women with chronic renal disease, which is associated with an increased risk for these adverse pregnancy outcomes. Due to the limited number of exposed pregnancies and multiple confounding factors (such as underlying maternal conditions, other maternal medications, and gestational timing of exposure), these published case reports and studies do not reliably estimate the frequency, presence or absence of adverse outcomes.
Animal Data
When rats received PROCRIT at doses greater than or equal to 100 Units/kg/day during mating and through early pregnancy (dosing stopped prior to organogenesis), there were slight increases in the incidences of pre- and post-implantation loss, and a decrease in live fetuses in the presence of maternal toxicity (red limbs/pinna, focal splenic capsular toxicity, increased organ weights). This animal dose level of 100 Units/kg/day may approximate the clinical recommended starting dose, depending on the treatment indication. When pregnant rats and rabbits received intravenous doses of up to 500 mg/kg/day of PROCRIT only during organogenesis (gestational days 7 to 17 in rats and gestational days 6 to 18 in rabbits), no teratogenic effects were observed in the offspring. The offspring (F1 generation) of the treated rats were observed postnatally; rats from the F1 generation reached maturity and were mated; no PROCRIT-related effects were apparent for their offspring (F2 generation fetuses).
When pregnant rats received PROCRIT at doses of 500 Units/kg/day late in pregnancy (after the period of organogenesis from day 17 of gestation through day 21 of lactation), pups exhibited decreased number of caudal vertebrae, decreased body weight gain, and delayed appearance of abdominal hair, eyelid opening, and ossification in the presence of maternal toxicity (red limbs/pinna, increased organ weights). This animal dose level of 500 U/kg/day is approximately five times the clinical recommended starting dose depending on the patient's treatment indication.
8.2 Lactation
Risk Summary
PROCRIT from multiple-dose vials contains benzyl alcohol and is contraindicated in lactating women [see Contraindications (4), Warnings and Precautions (5.9)] . Advise a lactating woman not to breastfeed for at least 2 weeks after the last dose. The preservative benzyl alcohol has been associated with serious adverse reactions and death when administered intravenously to neonates and infants [see Use in Specific Populations (8.4)]. There is a potential for similar risks to infants exposed to benzyl alcohol through human milk.
Do not mix PROCRIT with bacteriostatic saline containing benzyl alcohol, if administering PROCRIT to a lactating woman [see Dosage and Administration (2.1)] .
There is no information regarding the presence of PROCRIT in human milk, the effects on the breastfed infant, or the effects on milk production. However, endogenous erythropoietin is present in human milk. Because many drugs are present in human milk, caution should be exercised when PROCRIT from single-dose vials is administered to a lactating woman.
8.4 Pediatric Use
The multiple-dose vials are formulated with benzyl alcohol and are contraindicated for use in neonates and infants [see Contraindications (4), Warnings and Precautions (5.9)] . When therapy with PROCRIT is needed in neonates and infants, use the single-dose vial, which is a benzyl alcohol-free formulation. Do not mix the single-dose vials with bacteriostatic saline when administering PROCRIT to neonates or infants because it contains benzyl alcohol [see Dosage and Administration (2.6)] .
Serious adverse reactions including fatal reactions and the "gasping syndrome" occurred in premature neonates and infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.378 mmol/L). Additional adverse reactions included gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Preterm, low birth weight infants may be more likely to develop these reactions because they may be less able to metabolize benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known [see Warnings and Precautions (5.9)] .
Pediatric Patients with CKD
PROCRIT is indicated in pediatric patients, ages 1 month to 16 years of age, for the treatment of anemia associated with CKD requiring dialysis. Safety and effectiveness in pediatric patients less than 1 month old have not been established [see Clinical Studies (14.1)] .
Use of PROCRIT in pediatric patients with CKD not requiring dialysis is supported by efficacy in pediatric patients requiring dialysis. The mechanism of action of PROCRIT is the same for these two populations. Published literature also has reported the use of PROCRIT in pediatric patients with CKD not requiring dialysis. Dose-dependent increases in hemoglobin and hematocrit were observed with reductions in transfusion requirements.
The safety data from the pediatric studies and postmarketing reports are similar to those obtained from the studies of PROCRIT in adult patients with CKD [see Warnings and Precautions (5)and Adverse Reactions (6.1)] . Postmarketing reports do not indicate a difference in safety profiles in pediatric patients with CKD requiring dialysis and not requiring dialysis.
Pediatric Patients with Cancer on Chemotherapy
PROCRIT is indicated in patients 5 to 18 years old for the treatment of anemia due to concomitant myelosuppressive chemotherapy. Safety and effectiveness in pediatric patients less than 5 years of age have not been established [see Clinical Studies (14.3)]. The safety data from these studies are similar to those obtained from the studies of PROCRIT in adult patients with cancer [see Warnings and Precautions (5.1, 5.2)and Adverse Reactions (6.1)].
Pediatric Patients with HIV Infection Receiving Zidovudine
Published literature has reported the use of PROCRIT in 20 zidovudine-treated, anemic, pediatric patients with HIV Infection, ages 8 months to 17 years, treated with 50 to 400 Units/kg subcutaneously or intravenously 2 to 3 times per week. Increases in hemoglobin levels and in reticulocyte counts and decreases in or elimination of RBC transfusions were observed.
Pharmacokinetics in Neonates
Limited pharmacokinetic data from a study of 7 preterm, very low birth weight neonates and 10 healthy adults given intravenous erythropoietin suggested that distribution volume was approximately 1.5 to 2 times higher in the preterm neonates than in the healthy adults, and clearance was approximately 3 times higher in the preterm neonates than in the healthy adults.
8.5 Geriatric Use
Of the 4553 patients who received PROCRIT in the 6 studies for treatment of anemia due to CKD not receiving dialysis, 2726 (60%) were age 65 years and over, while 1418 (31%) were 75 years and over. Of the 757 patients who received PROCRIT in the 3 studies of CKD patients on dialysis, 361 (47%) were age 65 years and over, while 100 (13%) were 75 years and over. No differences in safety or effectiveness were observed between geriatric and younger patients. Dose selection and adjustment for an elderly patient should be individualized to achieve and maintain the target hemoglobin [see Dosage and Administration (2)] .
Among 778 patients enrolled in the 3 clinical studies of PROCRIT for the treatment of anemia due to concomitant chemotherapy, 419 received PROCRIT and 359 received placebo. Of the 419 who received PROCRIT, 247 (59%) were age 65 years and over, while 78 (19%) were 75 years and over. No overall differences in safety or effectiveness were observed between geriatric and younger patients. The dose requirements for PROCRIT in geriatric and younger patients within the 3 studies were similar.
Among 1731 patients enrolled in the 6 clinical studies of PROCRIT for reduction of allogeneic RBC transfusions in patients undergoing elective surgery, 1085 received PROCRIT and 646 received placebo or standard of care treatment. Of the 1085 patients who received PROCRIT, 582 (54%) were age 65 years and over, while 245 (23%) were 75 years and over. No overall differences in safety or effectiveness were observed between geriatric and younger patients. The dose requirements for PROCRIT in geriatric and younger patients within the 4 studies using the 3 times weekly schedule and 2 studies using the weekly schedule were similar.
Insufficient numbers of patients age 65 years or older were enrolled in clinical studies of PROCRIT for the treatment of patients treated with zidovudine for HIV Infection to determine whether they respond differently from younger patients .
-
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
Abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects. Abuse of PROCRIT may be seen in athletes for the effects on erythropoiesis. Abuse of drugs that increase erythropoiesis, such as PROCRIT, by healthy persons may lead to life-threatening cardiovascular complications (e.g., stroke, myocardial infarction, or thromboembolism) [see Warnings and Precautions (5.1)].
In animal studies, epoetin alfa did not distribute to the CNS nor produce behavioral effects that are consistent with CNS activity.
-
10 OVERDOSAGE
PROCRIT overdosage can cause hemoglobin levels above the desired level, which should be managed with discontinuation or reduction of PROCRIT dosage and/or with phlebotomy, as clinically indicated [see Clinical Pharmacology (12.2)] . Cases of severe hypertension have been observed following overdose with ESAs [see Warnings and Precautions (5.3)].
-
11 DESCRIPTION
Epoetin alfa is a 165-amino acid erythropoiesis-stimulating glycoprotein manufactured by recombinant DNA technology. It has a molecular weight of approximately 30,400 daltons and is produced by mammalian cells into which the human erythropoietin gene has been introduced. The product contains the identical amino acid sequence of isolated natural erythropoietin.
PROCRIT (epoetin alfa) injection for intravenous or subcutaneous administration is formulated as a sterile, clear, colorless liquid in vials in multiple formulations. Single-dose vials, formulated with an isotonic sodium chloride/sodium citrate-buffered solution, are supplied in multiple strengths. Each single-dose 1 mL vial contains 2,000, 3,000, 4,000, or 10,000 Units of epoetin alfa, Albumin (Human) (2.5 mg), citric acid (0.06 mg), sodium chloride (5.9 mg), and sodium citrate (5.8 mg) in Water for Injection, USP (pH 6.9 ± 0.3). Single-dose 1 mL vials formulated with an isotonic sodium chloride/sodium phosphate buffer contain 40,000 Units of epoetin alfa albumin (human) (2.5 mg), citric acid (0.0068 mg), sodium chloride (5.8 mg), sodium citrate (0.7 mg), sodium phosphate dibasic anhydrate (1.8 mg), and sodium phosphate monobasic monohydrate (1.2 mg) in Water for Injection, USP (pH 6.9 ± 0.3). Multiple-dose, 2 mL vials contain 10,000 Units epoetin alfa, albumin (human) (2.5 mg), benzyl alcohol (1%), sodium chloride (8.2 mg), citric acid (0.11 mg), and sodium citrate (1.3 mg) per 1 mL Water for Injection, USP (pH 6.1 ± 0.3). Multiple-dose 1 mL vials contain 20,000 Units epoetin alfa, albumin (human) (2.5 mg), benzyl alcohol (1%), sodium chloride (8.2 mg), citric acid (0.11 mg), and sodium citrate (1.3 mg), per 1 mL in Water for Injection, USP (pH 6.1 ± 0.3).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
PROCRIT stimulates erythropoiesis by the same mechanism as endogenous erythropoietin.
12.2 Pharmacodynamics
PROCRIT increases the reticulocyte count within 10 days of initiation, followed by increases in the RBC count, hemoglobin, and hematocrit, usually within 2 to 6 weeks. The rate of hemoglobin increase varies among patients and is dependent upon the dose of PROCRIT administered. For correction of anemia in hemodialysis patients, a greater biologic response is not observed at doses exceeding 300 Units/kg 3 times weekly.
12.3 Pharmacokinetics
In adult and pediatric patients with CKD, the elimination half-life (t 1/2) of plasma erythropoietin after intravenous administration of PROCRIT ranged from 4 to 13 hours. After subcutaneous administration, C maxwas achieved within 5 to 24 hours. The t 1/2in adult patients with serum creatinine greater than 3 mg/dL was similar between those not on dialysis and those maintained on dialysis. The pharmacokinetic data indicate no apparent difference in PROCRIT t 1/2among adult patients above or below 65 years of age.
A pharmacokinetic study comparing 150 Units/kg subcutaneous 3 times weekly to 40,000 Units subcutaneous weekly dosing regimen was conducted for 4 weeks in healthy subjects (n=12) and for 6 weeks in anemic cancer patients (n=32) receiving cyclic chemotherapy. There was no accumulation of serum erythropoietin after the 2 dosing regimens during the study period. The 40,000 Units weekly regimen had a higher C max(3- to 7-fold), longer T max(2- to 3-fold), higher AUC 0–168 h(2- to 3-fold) of erythropoietin and lower clearance (CL) (50%) than the 150 Units/kg 3 times weekly regimen. In anemic cancer patients, the average t 1/2was similar (40 hours with range of 16 to 67 hours) after both dosing regimens. After the 150 Units/kg 3 times weekly dosing, the values of T maxand CL were similar (13.3 ± 12.4 vs. 14.2 ± 6.7 hours, and 20.2 ± 15.9 vs. 23.6 ± 9.5 mL/hr/kg) between week 1 when patients were receiving chemotherapy (n=14) and week 3 when patients were not receiving chemotherapy (n=4). Differences were observed after the 40,000 Units weekly dosing with longer T max(38 ± 18 hours) and lower CL (9.2 ± 4.7 mL/hr/kg) during week 1 when patients were receiving chemotherapy (n=18) compared with those (22 ± 4.5 hours, 13.9 ± 7.6 mL/hr/kg, respectively) during week 3 when patients were not receiving chemotherapy (n=7).
The pharmacokinetic profile of PROCRIT in pediatric patients appeared similar to that of adults.
The pharmacokinetics of PROCRIT has not been studied in patients with HIV Infection.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of PROCRIT has not been evaluated.
PROCRIT was not mutagenic or clastogenic under the conditions tested: PROCRIT was negative in the in vitrobacterial reverse mutation assay (Ames test), in the in vitromammalian cell gene mutation assay (the hypoxanthine-guanine phosphoribosyl transferase [HGPRT] locus), in an in vitrochromosomal aberration assay in mammalian cells, and in the in vivomouse micronucleus assay.
When administered intravenously to male and female rats prior to and during mating, and to females through the beginning of implantation (up to gestational day 7; dosing stopped prior to the beginning of organogenesis), doses of 100 and 500 Units/kg/day of PROCRIT caused slight increases in pre-implantation loss, post-implantation loss and decreases in the incidence of live fetuses. It is not clear whether these effects reflect a drug effect on the uterine environment or on the conceptus. This animal dose level of 100 Units/kg/day approximates the clinical recommended starting dose, depending on the patient's treatment indication, but may be lower than the clinical dose in patients whose doses have been adjusted.
-
14 CLINICAL STUDIES
14.1 Patients with Chronic Kidney Disease
Adult Patients on Dialysis
Patients with chronic kidney disease on dialysis: ESA effects on rates of transfusion
In clinical studies of patients with CKD on dialysis, PROCRIT increased hemoglobin levels and decreased the need for RBC transfusion. Overall, more than 95% of patients were RBC transfusion-independent after receiving PROCRIT for 3 months. In clinical studies at starting doses of 50 to 150 Units/kg 3 times weekly, adult patients responded with an average rate of hemoglobin rise as presented in Table 8.
Table 8: Average Rate of Hemoglobin Rise in 2 Weeks Starting Dose
(3 Times Weekly Intravenously)Hemoglobin Increase in 2 Weeks 50 Units/kg 0.5 g/dL 100 Units/kg 0.8 g/dL 150 Units/kg 1.2 g/dL The safety and efficacy of PROCRIT were evaluated in 13 clinical studies involving intravenous administration to a total of 1010 patients on dialysis with anemia. Overall, more than 90% of the patients treated with PROCRIT experienced improvement in hemoglobin concentrations. In the 3 largest of these clinical studies, the median maintenance dose necessary to maintain the hemoglobin between 10 to 12 g/dL was approximately 75 Units/kg 3 times weekly. More than 95% of patients were able to avoid RBC transfusions. In the largest US multicenter study, approximately 65% of the patients received doses of 100 Units/kg 3 times weekly or less to maintain their hemoglobin at approximately 11.7 g/dL. Almost 10% of patients received a dose of 25 Units/kg or less, and approximately 10% received a dose of more than 200 Units/kg 3 times weekly to maintain their hemoglobin at this level.
In the Normal Hematocrit Study, the yearly transfusion rate was 51.5% in the lower hemoglobin group (10 g/dL) and 32.4% in the higher hemoglobin group (14 g/dL).
Other ESA trials
In a 26-week, double-blind, placebo-controlled study, 118 patients on dialysis with an average hemoglobin of approximately 7 g/dL were randomized to either PROCRIT or placebo. By the end of the study, average hemoglobin increased to approximately 11 g/dL in the PROCRIT-treated patients and remained unchanged in patients receiving placebo. PROCRIT-treated patients experienced improvements in exercise tolerance and patient-reported physical functioning at month 2 that were maintained throughout the study.
A multicenter, unit-dose study was also conducted in 119 patients receiving peritoneal dialysis who self-administered PROCRIT subcutaneously. Patients responded to PROCRIT administered subcutaneously in a manner similar to patients receiving intravenous administration.
Pediatric Patients with CKD on Dialysis
The safety and efficacy of PROCRIT were studied in a placebo-controlled, randomized study of 113 pediatric patients with anemia (hemoglobin ≤ 9 g/dL) undergoing peritoneal dialysis or hemodialysis. The initial dose of PROCRIT was 50 Units/kg intravenously or subcutaneously 3 times weekly. The dose of study drug was titrated to achieve either a hemoglobin of 10 to 12 g/dL or an absolute increase in hemoglobin of 2 g/dL over baseline.
At the end of the initial 12 weeks, a statistically significant rise in mean hemoglobin (3.1 g/dL vs. 0.3 g/dL) was observed only in the PROCRIT arm. The proportion of pediatric patients achieving a hemoglobin of 10 g/dL, or an increase in hemoglobin of 2 g/dL over baseline, at any time during the first 12 weeks was higher in the PROCRIT arm (96% vs. 58%). Within 12 weeks of initiating PROCRIT therapy, 92.3% of the pediatric patients were RBC transfusion independent as compared to 65.4% who received placebo. Among patients who received 36 weeks of PROCRIT, hemodialysis patients received a higher median maintenance dose [167 Units/kg/week (n=28) vs. 76 Units/kg/week (n=36)] and took longer to achieve a hemoglobin of 10 to 12 g/dL (median time to response 69 days vs. 32 days) than patients undergoing peritoneal dialysis.
Adult Patients with CKD Not Requiring Dialysis
Four clinical studies were conducted in patients with CKD not on dialysis involving 181 patients treated with PROCRIT. These patients responded to PROCRIT therapy in a manner similar to that observed in patients on dialysis. Patients with CKD not on dialysis demonstrated a dose-dependent and sustained increase in hemoglobin when PROCRIT was administered by either an intravenous or subcutaneous route, with similar rates of rise of hemoglobin when PROCRIT was administered by either route.
Patients with chronic kidney disease not on dialysis: ESA effects on rates of transfusion
In TREAT, a randomized, double-blind trial of 4038 patients with CKD and type 2 diabetes not on dialysis, a post-hoc analysis showed that the proportion of patients receiving RBC transfusions was lower in patients administered an ESA to target a hemoglobin of 13 g/dL compared to the control arm in which an ESA was administered intermittently if hemoglobin concentration decreased to less than 9 g/dL (15% versus 25%, respectively). In CHOIR, a randomized open-label study of 1432 patients with CKD not on dialysis, use of epoetin alfa to target a higher (13.5 g/dL) versus lower (11.3 g/dL) hemoglobin goal did not reduce the use of RBC transfusions. In each trial, no benefits occurred for the cardiovascular or end-stage renal disease outcomes. In each trial, the potential benefit of ESA therapy was offset by worse cardiovascular safety outcomes resulting in an unfavorable benefit-risk profile [see Warnings and Precautions (5.1)] .
ESA Effects on Rates of Death and Other Serious Cardiac Adverse Reactions
Three randomized outcome trials (Normal Hematocrit Study [NHS], Correction of Anemia with Epoetin Alfa in Chronic Kidney Disease [CHOIR], and Trial of Darbepoetin Alfa in Type 2 Diabetes and CKD [TREAT]) have been conducted in patients with CKD using Epogen/PROCRIT/Aranesp to target higher vs. lower hemoglobin levels. Though these trials were designed to establish a cardiovascular or renal benefit of targeting higher hemoglobin levels, in all 3 studies, patients randomized to the higher hemoglobin target experienced worse cardiovascular outcomes and showed no reduction in progression to ESRD. In each trial, the potential benefit of ESA therapy was offset by worse cardiovascular safety outcomes resulting in an unfavorable benefit-risk profile [see Warnings and Precautions (5.1)] .
14.2 Zidovudine-treated Patients with HIV Infection
The safety and efficacy of PROCRIT were evaluated in 4 placebo-controlled studies enrolling 297 anemic patients (hemoglobin < 10 g/dL) with HIV Infection receiving concomitant therapy with zidovudine. In the subgroup of patients (89/125 PROCRIT and 88/130 placebo) with pre-study endogenous serum erythropoietin levels ≤ 500 mUnits/mL, PROCRIT reduced the mean cumulative number of units of blood transfused per patient by approximately 40% as compared to the placebo group. Among those patients who required RBC transfusions at baseline, 43% of patients treated with PROCRIT versus 18% of placebo-treated patients were RBC transfusion-independent during the second and third months of therapy. PROCRIT therapy also resulted in significant increases in hemoglobin in comparison to placebo. When examining the results according to the weekly dose of zidovudine received during month 3 of therapy, there was a statistically significant reduction (p < 0.003) in RBC transfusion requirements in patients treated with PROCRIT (n=51) compared to placebo-treated patients (n=54) whose mean weekly zidovudine dose was ≤ 4200 mg/week.
Approximately 17% of the patients with endogenous serum erythropoietin levels ≤ 500 mUnits/mL receiving PROCRIT in doses from 100 to 200 Units/kg 3 times weekly achieved a hemoglobin of 12.7 g/dL without administration of RBC transfusions or significant reduction in zidovudine dose. In the subgroup of patients whose pre-study endogenous serum erythropoietin levels were > 500 mUnits/mL, PROCRIT therapy did not reduce RBC transfusion requirements or increase hemoglobin compared to the corresponding responses in placebo-treated patients.
14.3 Patients with Cancer on Chemotherapy
The safety and effectiveness of PROCRIT was assessed in two multicenter, randomized (1:1), placebo-controlled, double-blind studies (Study C1 and Study C2) and a pooled analysis of six additional randomized (1:1), multicenter, placebo-controlled, double-blind studies. All studies were conducted in patients with anemia due to concomitantly administered cancer chemotherapy. Study C1 enrolled 344 adult patients, Study C2 enrolled 222 pediatric patients, and the pooled analysis contained 131 patients randomized to epoetin alfa or placebo. In Studies C1 and C2, efficacy was demonstrated by a reduction in the proportion of patients who received an RBC transfusion, from week 5 through end of the study, with the last-known RBC transfusion status carried forward for patients who discontinued treatment. In the pooled analysis, efficacy was demonstrated by a reduction in the proportion of patients who received an RBC transfusion from week 5 through end of the study in the subset of patients who were remaining on therapy for 6 or more weeks.
Study C1
Study C1 was conducted in patients with anemia (hemoglobin < 11.5 g/dL for males; < 10.5 g/dL for females) with non-myeloid malignancies receiving myelosuppressive chemotherapy. Randomization was stratified by type of malignancy (lung vs. breast vs. other), concurrent radiation therapy planned (yes or no), and baseline hemoglobin (< 9 g/dL vs. ≥ 9 g/dL); patients were randomized to epoetin alfa 40,000 Units (n=174) or placebo (n=170) as a weekly subcutaneous injection commencing on the first day of the chemotherapy cycle.
Ninety-one percent of patients were white, 44% were male, and the median age of patients was 66 years (range: 20 to 88 years). The proportion of patients withdrawn from the study prior to week 5 was less than 10% for placebo-treated or epoetin-treated patients. Per protocol, the last available hemoglobin values from patients who dropped out were included in the efficacy analyses. Efficacy results are shown in Table 9.
Study C2
Study C2 was conducted in 222 patients with anemia, ages 5 to 18, receiving chemotherapy for the treatment of various childhood malignancies. Randomization was stratified by cancer type (solid tumors, Hodgkin's disease, acute lymphocytic leukemia, vs. non-Hodgkin's lymphoma); patients were randomized to receive epoetin alfa at 600 Units/kg maximum 40,000 Units (n=111) or placebo (n=111) as a weekly intravenous injection.
Sixty-nine percent of patients were white, 55% were male, and the median age of patients was 12 years (range: 5 to 18 years). Two (2%) of placebo-treated patients and 3 (3%) of epoetin alfa-treated patients dropped out of the study prior to week 5. There were fewer RBC transfusions from week 5 through the end-of-study in epoetin-alfa treated patients [51% (57/111)] compared to placebo-treated patients [69% (77/111)]. There was no evidence of an improvement in health-related quality of life, including no evidence of an effect on fatigue, energy, or strength in patients receiving PROCRIT as compared to those receiving placebo.
Pooled Analysis (Three Times Per Week Dosing)
The results of 6 studies of similar design and that randomized 131 patients to epoetin alfa or placebo were pooled to assess the safety and effectiveness of epoetin alfa. Patients were randomized to receive epoetin alfa at 150 Units/kg (n=63) or placebo (n=68), subcutaneously three times per week for 12 weeks in each study. Across all studies, 72 patients were treated with concomitant non cisplatin-containing chemotherapy regimens and 59 patients were treated with concomitant cisplatin-containing chemotherapy regimens. Twelve patients (19%) in the epoetin alfa arm and 10 patients (15%) in the placebo-arm dropped out prior to week 6 and are excluded from efficacy analyses.
Table 10: Proportion of Patients Transfused in the Pooled Analysis for Three Times Per Week Dosing Week 5 Through Week 12 or End of Study * Chemotherapy Regimen PROCRIT Placebo All Regimens 22% (11/51) † 43% (25/58) Regimens without cisplatin 21% (6/29) 33% (11/33) Regimens containing cisplatin 23% (5/22) 56% (14/25) 14.4 Surgery Patients
The safety and efficacy of PROCRIT were evaluated in a placebo-controlled, double-blind study (S1) enrolling 316 patients scheduled for major, elective orthopedic hip or knee surgery who were expected to require ≥ 2 units of blood and who were not able or willing to participate in an autologous blood donation program. Patients were stratified into 1 of 3 groups based on their pretreatment hemoglobin [≤ 10 g/dL (n=2), > 10 to ≤ 13 g/dL (n=96), and > 13 to ≤ 15 g/dL (n=218)] and then randomly assigned to receive 300 Units/kg PROCRIT, 100 Units/kg PROCRIT, or placebo by subcutaneous injection for 10 days before surgery, on the day of surgery, and for 4 days after surgery. All patients received oral iron and a low-dose, postoperative warfarin regimen.
Treatment with PROCRIT 300 Units/kg significantly (p=0.024) reduced the risk of allogeneic RBC transfusion in patients with a pretreatment hemoglobin of > 10 to ≤ 13 g/dL; 5/31 (16%) of patients treated with PROCRIT 300 Units/kg, 6/26 (23%) of patients treated with PROCRIT 100 Units/kg, and 13/29 (45%) of placebo-treated patients were transfused. There was no significant difference in the number of patients transfused between PROCRIT (9% 300 Units/kg, 6% 100 Units/kg) and placebo (13%) in the > 13 to ≤ 15 g/dL hemoglobin stratum. There were too few patients in the ≤ 10 g/dL group to determine if PROCRIT is useful in this hemoglobin strata. In the > 10 to ≤ 13 g/dL pretreatment stratum, the mean number of units transfused per PROCRIT-treated patient (0.45 units blood for 300 Units/kg, 0.42 units blood for 100 Units/kg) was less than the mean transfused per placebo-treated patient (1.14 units) (overall p=0.028). In addition, mean hemoglobin, hematocrit, and reticulocyte counts increased significantly during the presurgery period in patients treated with PROCRIT.
PROCRIT was also evaluated in an open-label, parallel-group study (S2) enrolling 145 patients with a pretreatment hemoglobin level of ≥ 10 to ≤ 13 g/dL who were scheduled for major orthopedic hip or knee surgery and who were not participating in an autologous program. Patients were randomly assigned to receive 1 of 2 subcutaneous dosing regimens of PROCRIT (600 Units/kg once weekly for 3 weeks prior to surgery and on the day of surgery, or 300 Units/kg once daily for 10 days prior to surgery, on the day of surgery, and for 4 days after surgery). All patients received oral iron and appropriate pharmacologic anticoagulation therapy.
From pretreatment to presurgery, the mean increase in hemoglobin in the 600 Units/kg weekly group (1.44 g/dL) was greater than that observed in the 300 Units/kg daily group. The mean increase in absolute reticulocyte count was smaller in the weekly group (0.11 × 10 6/mm 3) compared to the daily group (0.17 × 10 6/mm 3). Mean hemoglobin levels were similar for the 2 treatment groups throughout the postsurgical period.
The erythropoietic response observed in both treatment groups resulted in similar RBC transfusion rates [11/69 (16%) in the 600 Units/kg weekly group and 14/71 (20%) in the 300 Units/kg daily group]. The mean number of units transfused per patient was approximately 0.3 units in both treatment groups.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
PROCRIT (epoetin alfa) injection is a sterile, clear, and colorless solution available as:
Preservative-free, single-dose vials (in citrate-buffered formulation): 2,000 Units/mL (NDC 59676-302-01), 3,000 Units/mL (NDC 59676-303-01), 4,000 Units/mL (NDC 59676-304-01), or 10,000 Units/mL (NDC 59676-310-01) supplied in cartons, each carton containing six 1 mL single-dose vials.
Preservative-free, single-dose vials (in citrate-buffered formulation): 10,000 Units/mL (NDC 59676-310-02) supplied in dispensing packs (tray) containing 25 single-dose 1 mL vials.
Preservative-free, single-dose vials (in phosphate-buffered formulation): 40,000 Units/mL (NDC 59676-340-01) supplied in dispensing packs containing four 1 mL single-dose vials.
Preserved, multiple-dose vials: 20,000 Units/2mL (10,000 Units/mL) (NDC 59676-312-04) supplied in dispensing packs containing four 2 mL multiple-dose vials.
Preserved, multiple-dose vials: 20,000 Units/mL (NDC 59676-320-04) supplied in dispensing packs containing four 1 mL multiple-dose vials.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Inform patients:
- Of the increased risks of mortality, serious cardiovascular reactions, thromboembolic reactions, stroke, and tumor progression [see Warnings and Precautions (5.1, 5.2)] .
- To undergo regular blood pressure monitoring, adhere to prescribed anti-hypertensive regimen and follow recommended dietary restrictions.
- To contact their healthcare provider for new-onset neurologic symptoms or change in seizure frequency.
- Of the need to have regular laboratory tests for hemoglobin.
- Risks are associated with benzyl alcohol in neonates, infants, pregnant women, and lactating women [see Use in Specific Populations (8.1, 8.2, 8.4)].
Instruct patients who self-administer PROCRIT of the:
- Importance of following the Instructions for Use.
- Dangers of reusing needles, syringes, or unused portions of single-dose vials.
- Proper disposal of used syringes, needles, and unused vials, and of the full container.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised 04/2024 MEDICATION GUIDE
PROCRIT ®(PRO'–KRIT)
(epoetin alfa)Read this Medication Guide:
- before you start PROCRIT.
- if you are told by your healthcare provider that there is new information about PROCRIT.
- if you are told by your healthcare provider that you may inject PROCRIT at home, read this Medication Guide each time you receive a new supply of medicine.
This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or your treatment. Talk with your healthcare provider regularly about the use of PROCRIT and ask if there is new information about PROCRIT.
What is the most important information I should know about PROCRIT?
PROCRIT may cause serious side effects that can lead to death, including:
For people with cancer:
- Your tumor may grow faster and you may die sooner if you choose to take PROCRIT. Your healthcare provider will talk with you about these risks.
For all people who take PROCRIT, including people with cancer or chronic kidney disease:
- Serious heart problems, such as heart attack or heart failure, and stroke.You may die sooner if you are treated with PROCRIT to increase red blood cells (RBCs) to near the same level found in healthy people.
- Blood clots.Blood clots may happen at any time while taking PROCRIT. If you are receiving PROCRIT for any reason and you are going to have surgery, talk to your healthcare provider about whether or not you need to take a blood thinner to lessen the chance of blood clots during or following surgery. Blood clots can form in blood vessels (veins), especially in your leg (deep venous thrombosis or DVT). Pieces of a blood clot may travel to the lungs and block the blood circulation in the lungs (pulmonary embolus).
- Call your healthcare provider or get medical help right away if you have any of these symptoms:
- Chest pain
- Trouble breathing or shortness of breath
- Pain in your legs, with or without swelling
- A cool or pale arm or leg
- Sudden confusion, trouble speaking, or trouble understanding others' speech
- Sudden numbness or weakness in your face, arm, or leg, especially on one side of your body
- Sudden trouble seeing
- Sudden trouble walking, dizziness, loss of balance or coordination
- Loss of consciousness (fainting)
- Hemodialysis vascular access stops working
See " What are the possible side effects of PROCRIT?" below for more information.
If you decide to take PROCRIT, your healthcare provider should prescribe the smallest dose of PROCRIT that is necessary to reduce your chance of needing RBC transfusions.
What is PROCRIT?
PROCRIT is a prescription medicine used to treat anemia. People with anemia have a lower-than-normal number of RBCs. PROCRIT works like the human protein called erythropoietin to help your body make more RBCs.
PROCRIT is used to reduce or avoid the need for RBC transfusions.
PROCRIT may be used to treat anemia if it is caused by:
- Chronic kidney disease (you may or may not be on dialysis).
- Chemotherapy that will be used for at least two months after starting PROCRIT.
- A medicine called zidovudine (AZT) used to treat HIV infection.
PROCRIT may also be used to reduce the chance you will need RBC transfusions if you are scheduled for certain surgeries where a lot of blood loss is expected.
If your hemoglobin level stays too high or if your hemoglobin goes up too quickly, this may lead to serious health problems which may result in death. These serious health problems may happen if you take PROCRIT, even if you do not have an increase in your hemoglobin level.
PROCRIT has not been proven to improve quality of life, fatigue, or well-being.
PROCRIT should not be usedfor treatment of anemia:
- If you have cancer and you will not be receiving chemotherapy that may cause anemia.
- If you have a cancer that has a high chance of being cured. Talk with your healthcare provider about the kind of cancer you have.
- If your anemia caused by chemotherapy treatment can be managed by RBC transfusion.
- In place of emergency treatment for anemia (RBC transfusions).
PROCRIT should not be used to reduce the chance you will need RBC transfusions if:
- You are scheduled for surgery on your heart or blood vessels.
- You are able and willing to donate blood prior to surgery.
It is not known if PROCRIT is safe and effective in treating anemia in children less than 1 month old who have chronic kidney disease and in children less than 5 years old who have anemia caused by chemotherapy.
Do not take PROCRIT if you:
- Have cancer and have not been counseled by your healthcare provider about treatment with PROCRIT.
- Have high blood pressure that is not controlled (uncontrolled hypertension).
- Have been told by your healthcare provider that you have or have ever had a type of anemia called Pure Red Cell Aplasia (PRCA) that starts after treatment with PROCRIT or other erythropoietin protein medicines.
- Have had a serious allergic reaction to PROCRIT.
Do notgive PROCRIT from multiple-dose vials to:
- Pregnant or breastfeeding women
- Babies
Before taking PROCRIT, tell your healthcare provider about all of your medical conditions,including if you:
- Have heart disease.
- Have high blood pressure.
- Have had a seizure (convulsion) or stroke.
- Receive dialysis treatment.
- Are pregnant or plan to become pregnant. It is not known if PROCRIT may harm your unborn baby. Talk to your healthcare provider about possible pregnancy and birth control choices that are right for you.
- Are breastfeeding or plan to breastfeed. It is not known if PROCRIT passes into breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I take PROCRIT?
- If you or your caregiver has been trained to give PROCRIT shots (injections) at home:
- Be sure that you read, understand, and follow the "Instructions for Use" that come with PROCRIT.
- Take PROCRIT exactly as your healthcare provider tells you to. Do not change the dose of PROCRIT unless told to do so by your healthcare provider.
- Your healthcare provider will show you how much PROCRIT to use, how to inject it, how often it should be injected, and how to safely throw away the used vials, syringes, and needles.
- If you miss a dose of PROCRIT, call your healthcare provider right away and ask what to do.
- If you take more than the prescribed dose of PROCRIT, call your healthcare provider right away.
- During treatment with PROCRIT, continue to follow your healthcare provider's instructions for diet and medicines.
- Have your blood pressure checked as instructed by your healthcare provider.
What are the possible side effects of PROCRIT?
PROCRIT may cause serious side effects, including:
- See " What is the most important information I should know about PROCRIT?"
- High blood pressure.High blood pressure is a common side effect of PROCRIT in people with chronic kidney disease. Your blood pressure may go up or be difficult to control with blood pressure medicine while taking PROCRIT. This can happen even if you have never had high blood pressure before. Your healthcare provider should check your blood pressure often. If your blood pressure does go up, your healthcare provider may prescribe new or more blood pressure medicine.
- Seizures.If you have any seizures while taking PROCRIT, get medical help right away and tell your healthcare provider.
- Antibodies to PROCRIT.Your body may make antibodies to PROCRIT. These antibodies can block or lessen your body's ability to make RBCs and cause you to have severe anemia. Call your healthcare provider if you have unusual tiredness, lack of energy, dizziness, or fainting. You may need to stop taking PROCRIT.
- Serious allergic reactions.Serious allergic reactions can cause a skin rash, itching, shortness of breath, wheezing, dizziness and fainting because of a drop in blood pressure, swelling around your mouth or eyes, fast pulse, or sweating. If you have a serious allergic reaction, stop using PROCRIT and call your healthcare provider or get medical help right away.
- Severe skin reactions.Signs and symptoms of severe skin reactions with PROCRIT may include: skin rash with itching, blisters, skin sores, peeling, or areas of skin coming off. If you have any signs or symptoms of a severe skin reaction, stop using PROCRIT and call your healthcare provider or get medical help right away.
- Dangers of using PROCRIT from multiple-dose vials in newborns, infants, and pregnant or breastfeeding women.Do not use PROCRIT from multiple-dose vials in newborns, infants, pregnant or breastfeeding women because the PROCRIT in these vials contains benzyl alcohol. Benzyl alcohol has been shown to cause brain damage, other serious side effects, and death in newborn and premature babies. If you use PROCRIT from multiple-dose vials you should not breastfeed for at least 2 weeks after the last dose. PROCRIT that comes in single-dose vials does not contain benzyl alcohol. See " Who should not take PROCRIT?"
Common side effects of PROCRIT include:
- joint, muscle, or bone pain
- fever
- cough
- dizziness
- high blood sugar
- low potassium levels in the blood
- chills
- rash
- nausea
- vomiting
- blood vessel blockage
- low white blood cells
- trouble sleeping
- difficulty swallowing
- soreness of mouth
- itching
- headache
- respiratory infection
- weight decrease
- depression
- muscle spasm
- redness and pain at the PROCRIT injection site
These are not all of the possible side effects of PROCRIT. Your healthcare provider can give you a more complete list. Tell your healthcare provider about any side effects that bother you or that do not go away.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store PROCRIT?
- Do not shake PROCRIT.
- Store PROCRIT vials in the carton it comes in to protect from light.
- Store PROCRIT in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Do not freeze PROCRIT.Do not use PROCRIT that has been frozen.
- Throw away multiple-dose vials of PROCRIT no later than 21 days from the first day that you put a needle into the vial.
- Single-dose vials of PROCRIT should be used only one time. Throw the vial away after use even if there is medicine left in the vial.
Keep PROCRIT and all medicines out of the reach of children.
General information about the safe and effective use of PROCRIT
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use PROCRIT for a condition for which it was not prescribed. Do not give PROCRIT to other people even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about PROCRIT that is written for healthcare professionals.
What are the ingredients in PROCRIT?
Active ingredient:epoetin alfa
Inactive ingredients:
- Multiple-dose vials contain benzyl alcohol.
- All vials contain albumin (human), citric acid, sodium chloride, sodium citrate and Water for Injection.
- Single-dose vials containing 40,000 Units of PROCRIT also contain sodium phosphate dibasic anhydrate and sodium phosphate monobasic monohydrate.
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799 U.S.A.Manufactured for:
Janssen Products, LP
Horsham, Pennsylvania 19044 U.S.A.
© 2000 Janssen Pharmaceutical CompaniesFor more information, go to the following website: www.PROCRIT.com or call 1-800-JANSSEN (1-800-526-7736).
-
INSTRUCTIONS FOR USE
Instructions for Use
PROCRIT ®(PRO'–KRIT)
(epoetin alfa)Use these Instructions for Use if you or your caregiver has been trained to give PROCRIT injections at home. Do not give yourself the injection unless you have received training from your healthcare provider. If you are not sure about giving the injection or you have questions, ask your healthcare provider for help.
Before reading these Instructions for Use, read the Medication Guide that comes with PROCRIT for the most important information you need to know.
When you receive your PROCRIT vial and syringes make sure that:
- The name PROCRIT appears on the carton and vial label.
- The expiration date on the vial label has not passed. Do not use a vial of PROCRIT after the expiration date on the label.
- The dose strength of the PROCRIT vial (number of Units per mL on the vial label) is the same as your healthcare provider prescribed.
- You understand what the dose strength of PROCRIT means. PROCRIT vials come in several dose strengths. For example, the dose strength may be described as 10,000 Units/mL on the vial label. This strength means that 10,000 Units of medicine are contained in each 1 mL (milliliter) of liquid. Your healthcare provider may also refer to a mL as a "cc." One mL is the same as one "cc."
- The PROCRIT liquid in the vial is clear and colorless. Do not use PROCRIT if the liquid in the vial looks discolored or cloudy, or if the liquid has lumps, flakes, or particles.
- The PROCRIT vial has a color cap on the top of the vial. Do not use a vial of PROCRIT if the color cap on the top of the vial has been removed or is missing.
- Use only the type of disposable syringe and needle that your healthcare provider has prescribed.
- Do not shake PROCRIT. Shaking could cause PROCRIT not to work. If you shake PROCRIT, the solution in the vial may look foamy and should not be used.
- Do not freeze PROCRIT. Do not use a vial of PROCRIT that has been frozen.
- Store PROCRIT in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep PROCRIT away from light.
- Single-dose vials of PROCRIT should be used only one time. Throw the vial away after use even if there is medicine left in the vial.
- After removing a dose from the multidose vial, store the vial in the refrigerator (but not the freezer). Do not store the vial for more than 21 days.
- Throw away the multidose vial as directed by your healthcare provider:
- if there is not enough medicine left in the multidose vial for another dose, or
- if it has been more than 21 days since you first put a needle into the multidose vial.
How should I prepare for an injection of PROCRIT?
- Always keep an extra syringe and needle on hand.
- Follow your healthcare provider's instructions on how to measure your dose of PROCRIT. This dose will be measured in Units per mL or cc (1 mL is the same as 1 cc). Use a syringe that is marked in tenths of mL (for example, 0.2 mL or 0.2 cc). Using the wrong syringe can lead to a mistake in your dose and you could inject too much or too little PROCRIT.
Only use disposable syringes and needles. Use the syringes and needles only one time and then throw them away as instructed by your healthcare provider.
What do I need to know about the different types of PROCRIT vials?
PROCRIT comes in two different types of vials.
- Single-dose Vials
- Multidose Vials
The multidose vial of PROCRIT contains the preservative benzyl alcohol. Benzyl alcohol has been shown to cause brain damage, other serious side effects, and death in newborn and premature babies.PROCRIT that comes in single-dose vials does not contain benzyl alcohol.
Important: Follow these instructions exactly to help avoid infections.
Preparing the dose:
- Remove the vial of PROCRIT from the refrigerator. During this time, protect the solution from light.
- Do not use a single-dose vial of PROCRIT more than one time.
- Do not shake PROCRIT.
- Gather the other supplies you will need for your injection (vial, syringe, alcohol wipes, cotton ball, and a puncture-proof container for throwing away the syringe and needle). See Figure 1.
- Check the date on the PROCRIT vial to be sure that the drug has not expired.
- Wash your hands well with soap and water before preparing the medicine. See Figure 2.
- Flip off the protective color cap on the top of the vial. Do not remove the grey rubber stopper. Wipe the top of the grey rubber stopper with an alcohol wipe. See Figures 3and 4.
Figure 3 Figure 4 - Check the package containing the syringe. If the package has been opened or damaged, do not use that syringe. Throw away the syringe in the puncture-proof disposable container. If the syringe package is undamaged, open the package and remove the syringe.
- Using a syringe and needle that has been recommended by your healthcare provider, carefully remove the needle cover. See Figure 5. Then draw air into the syringe by pulling back on the plunger. The amount of air drawn into the syringe should be equal to the amount (mL or cc) of the PROCRIT dose prescribed by your healthcare provider. See Figure 6.
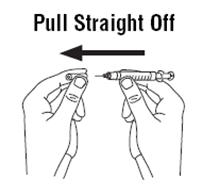
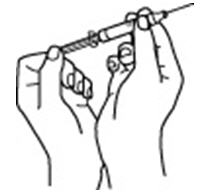
Figure 5 Figure 6 - With the vial on a flat work surface, insert the needle straight down through the grey rubber stopper of the PROCRIT vial. See Figure 7.
- Push the plunger of the syringe down to inject the air from the syringe into the vial of PROCRIT. The air injected into the vial will allow PROCRIT to be easily withdrawn into the syringe. See Figure 7.
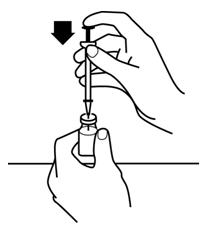
- Keep the needle inside the vial. Turn the vial and syringe upside down. Be sure the tip of the needle is in the PROCRIT liquid. Keep the vial upside down. Slowly pull back on the plunger to fill the syringe with PROCRIT liquid to the number (mL or cc) that matches the dose your healthcare provider prescribed. See Figure 8.
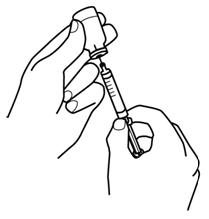
- Keep the needle in the vial. Check for air bubbles in the syringe. A small amount of air is harmless. Too large an air bubble will give you the wrong PROCRIT dose. To remove air bubbles, gently tap the syringe with your fingers until the air bubbles rise to the top of the syringe. Slowly push the plunger up to force the air bubbles out of the syringe. Keep the tip of the needle in the PROCRIT liquid. Pull the plunger back to the number on the syringe that matches your dose. Check again for air bubbles. If there are still air bubbles, repeat the steps above to remove them. See Figures 9and 10.
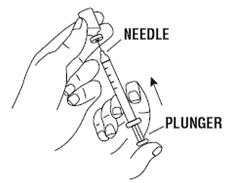
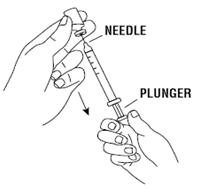
Figure 9 Figure 10 - Double-check that you have the correct dose in the syringe. Lay the vial down on its side with the needle still in it until after you have selected and prepared your site for injection.
Selecting and preparing the injection site:
PROCRIT can be injected into your body using two different ways (routes) as described below. Follow your healthcare provider's instructions about how you should inject PROCRIT. In patients on hemodialysis, the intravenous (IV) route is recommended.
-
Subcutaneous Route:
- PROCRIT can be injected directly into a layer of fat under your skin. This is called a subcutaneous injection. When giving subcutaneous injections, follow your healthcare provider's instructions about changing the site for each injection. You may wish to write down the site where you have injected.
- Do not inject PROCRIT into an area that is tender, red, bruised, hard, or has scars or stretch marks. Recommended sites for injection are shown in Figure 11 below, including:
- The outer area of the upper arms
- The abdomen (except for the 2-inch area around the navel)
- The front of the middle thighs
- The upper outer area of the buttocks
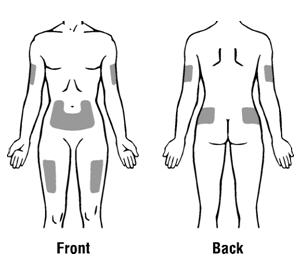
- Clean the skin with an alcohol wipe where the injection is to be made. Be careful not to touch the skin that has been wiped clean. See
Figure 12.
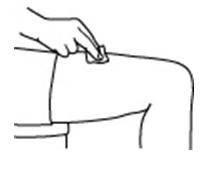
- Double-check that the correct amount of PROCRIT is in the syringe.
- Remove the prepared syringe and needle from the vial of PROCRIT and hold it in the hand that you will use to inject the medicine.
- Use the other hand to pinch a fold of skin at the cleaned injection site. Do not touch the cleaned area of skin. See
Figure 13.
- Hold the syringe like you would hold a pencil. Use a quick "dart-like" motion to insert the needle either straight up and down (90-degree angle) or at a slight angle (45 degrees) into the skin. Inject the prescribed dose subcutaneously as directed by your doctor, nurse or pharmacist. See
Figure 14.
- Pull the needle out of the skin and press a cotton ball or gauze over the injection site and hold it there for several seconds. Do not recap the needle.
- Dispose of the used syringe and needle as described below. Do not reuse syringes and needles.
-
Intravenous Route:
- PROCRIT can be injected in your vein through a special access port placed by your healthcare provider. This type of PROCRIT injection is called an intravenous (IV) injection. This route is usually for hemodialysis patients.
- If you have a dialysis vascular access, make sure it is working by checking it as your healthcare provider has shown you. Be sure to let your healthcare provider know right away if you are having any problems, or if you have any questions.
- Wipe off the venous port of the hemodialysis tubing with an alcohol wipe. See
Figure 15.
- Insert the needle of the syringe into the cleaned venous port and push the plunger all the way down to inject all the PROCRIT. See
Figure 16.
- Remove the syringe from the venous port. Do not recap the needle.
- Dispose of the used syringe and needle as described below.
How should I dispose of the vials, syringes, and needles?
Do not reuse the single-dose vials, syringes, or needles. Throw away the vials, syringes, and needles as instructed by your healthcare provider or by following these steps:
- Do not throw the vials, syringes, or needles in the household trash or recycle.
- Do not put the needle cover back on the needle.
- Place all used needles and syringes in a puncture-proof disposable container with a lid. Do not use glass or clear plastic containers, or any container that will be recycled or returned to a store.
- Keep the puncture-proof disposable container out of the reach of children.
- When the puncture-proof disposable container is full, tape around the cap or lid to make sure the cap or lid does not come off. Throw away the puncture-proof disposable container as instructed by your healthcare provider. There may be special state and local laws for disposing of used needles and syringes. Do not throw the puncture-proof disposable container in the household trash. Do not recycle.
Keep PROCRIT and all medicines out of reach of children.
These Instructions for Use have been approved by the U.S. Food and Drug Administration.
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799 U.S.A.Manufactured for:
Janssen Products, LP
Horsham, Pennsylvania 19044
© Janssen Products, LP 2000
Printed in U. S. A.Revised: 05/2012
-
PRINCIPAL DISPLAY PANEL - 2,000 Unit Vial Carton
2,000 Units/mL
NDC59676 -302-01
6-1 mL Single-dose vials
Discard unused portion.PROCRIT ®
EPOETIN ALFA
2,000 Units/mLFor Intravenous or Subcutaneous Use Only
Sterile Solution — No PreservativeAttention: A medication
guide is required for each patient.
For more copies see Procrit.com
or call 1-800-JANSSEN (1-800-526-7736)Each 1 mL vial contains: 2,000 units of recombinant epoetin alfa and
2.5 mg Albumin (Human) in a sterile, buffered solution (pH 6.9 ± 0.3)
of sodium citrate (5.8 mg), sodium chloride (5.9 mg) and citric acid
(0.06 mg) in Water for Injection, USP. No U.S. standard of potency.Rx only

-
PRINCIPAL DISPLAY PANEL - 3,000 Unit Vial Carton
3,000 Units/mL
NDC59676 -303-01
6-1 mL Single-dose vials
Discard unused portion.PROCRIT ®
EPOETIN ALFA
3,000 Units/mLFor Intravenous or Subcutaneous Use Only
Sterile Solution — No PreservativeAttention: A medication
guide is required for each patient.
For more copies see Procrit.com
or call 1-800-JANSSEN (1-800-526-7736)Each 1 mL vial contains: 3,000 units of recombinant epoetin alfa and
2.5 mg Albumin (Human) in a sterile, buffered solution (pH 6.9 ± 0.3)
of sodium citrate (5.8 mg), sodium chloride (5.9 mg) and citric acid
(0.06 mg) in Water for Injection, USP. No U.S. standard of potency.Rx only

-
PRINCIPAL DISPLAY PANEL - 4,000 Unit Vial Carton
4,000 Units/mL
NDC59676 -304-01
6-1 mL Single-dose vials
Discard unused portion.PROCRIT ®
EPOETIN ALFA
4,000 Units/mLFor Intravenous or Subcutaneous Use Only
Sterile Solution — No PreservativeAttention: A medication
guide is required for each patient.
For more copies see Procrit.com
or call 1-800-JANSSEN (1-800-526-7736)Each 1 mL vial contains: 4,000 units of recombinant epoetin alfa and
2.5 mg Albumin (Human) in a sterile, buffered solution (pH 6.9 ± 0.3)
of sodium citrate (5.8 mg), sodium chloride (5.9 mg) and citric acid
(0.06 mg) in Water for Injection, USP. No U.S. standard of potency.Rx only
-
PRINCIPAL DISPLAY PANEL - 10,000 Unit Vial Carton
10,000 Units/mL
NDC59676 -310-01
6-1 mL Single-dose vials
Discard unused portion.PROCRIT ®
EPOETIN ALFA
10,000 Units/mLFor Intravenous or Subcutaneous Use Only
Sterile Solution — No PreservativeAttention: A medication
guide is required for each patient.
For more copies see Procrit.com
or call 1-800-JANSSEN (1-800-526-7736)Each 1 mL vial contains: 10,000 units of recombinant epoetin alfa and
2.5 mg Albumin (Human) in a sterile, buffered solution (pH 6.9 ± 0.3)
of sodium citrate (5.8 mg), sodium chloride (5.9 mg) and citric acid
(0.06 mg) in Water for Injection, USP. No U.S. standard of potency.Rx only
-
PRINCIPAL DISPLAY PANEL - 40,000 Unit Vial Carton
40,000 Units/mL
NDC59676 -340-01
4-1 mL Single-dose vials
Discard unused portion.PROCRIT ®
EPOETIN ALFA
40,000 Units/mLFor Intravenous or Subcutaneous Use Only
Sterile Solution — No PreservativeAttention: A medication
guide is required for each patient.
For more copies see Procrit.com
or call 1-800-JANSSEN
(1-800-526-7736)Each 1 mL vial contains: 40,000 units of recombinant
epoetin alfa and 2.5 mg Albumin (Human) in a sterile,
buffered solution (pH 6.9 ± 0.3) of sodium phosphate
monobasic monohydrate (1.2 mg), sodium phosphate
dibasic anhydrate (1.8 mg), sodium citrate (0.7 mg),
sodium chloride (5.8 mg), and citric acid (6.8 mcg) in
Water for Injection, USP. No U.S. standard of potency.Rx only
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 20,000 Units/2 mL Vial Carton
20,000 Units/2 mL
(10,000 Units/mL)NDC59676-312-04
4-2 mL Multiple-dose VialsPROCRIT ®
EPOETIN ALFA20,000 Units/2 mL (10,000 Units/mL)
Multiple-dose Vials
Sterile solution — Preserved. 10,000 Units/mL
For Intravenous or Subcutaneous Use Only.Attention:
A medication guide is
required for each
patient. For more
copies see Procrit.com
or call 1-800-JANSSEN
(1-800-526-7736)2 mL
Rx only

-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL - 20,000 Unit Vial Carton
20,000 Units/mL
NDC59676-320-04
4-1 mL Multiple-dose VialsPROCRIT ®
EPOETIN ALFA20,000 Units/mL
Multiple-dose Vials
Sterile solution — Preserved. 20,000 Units/mL
For Intravenous or Subcutaneous Use OnlyAttention:
A medication guide is required
for each patient. For more copies
see Procrit.com or call
1-800-JANSSEN (1-800-526-7736)1 mL
Rx only

-
INGREDIENTS AND APPEARANCE
PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-303 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 3000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 5.8 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.06 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-303-01 6 in 1 CARTON 06/01/1989 1 NDC:59676-303-00 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-304 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 4000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 5.8 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.06 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-304-01 6 in 1 CARTON 06/01/1989 1 NDC:59676-304-00 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-310 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 10000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 5.8 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.06 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-310-01 6 in 1 CARTON 06/01/1989 1 NDC:59676-310-00 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:59676-310-02 25 in 1 CARTON 06/01/1989 2 NDC:59676-310-00 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-302 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 2000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 5.8 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.06 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-302-01 6 in 1 CARTON 06/01/1989 1 NDC:59676-302-00 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-340 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 40000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 1.2 mg in 1 mL SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 1.8 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 0.7 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 6.8 ug in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-340-01 4 in 1 CARTON 06/01/1989 1 NDC:59676-340-00 1 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-312 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 10000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 1.3 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 8.2 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.11 mg in 1 mL BENZYL ALCOHOL (UNII: LKG8494WBH) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-312-04 4 in 1 CARTON 06/01/1989 1 NDC:59676-312-00 2 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 PROCRIT
erythropoietin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:59676-320 Route of Administration INTRAVENOUS, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EPOETIN (UNII: 64FS3BFH5W) (EPOETIN - UNII:64FS3BFH5W) EPOETIN 20000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 2.5 mg in 1 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 5.8 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) 0.06 mg in 1 mL BENZYL ALCOHOL (UNII: LKG8494WBH) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:59676-320-04 4 in 1 CARTON 06/01/1989 1 NDC:59676-320-00 1 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103234 06/01/1989 Labeler - Janssen Products, LP (804684207) Establishment Name Address ID/FEI Business Operations Amgen Manufacturing Ltd 785800020 manufacture(59676-302, 59676-303, 59676-304, 59676-310, 59676-340, 59676-312, 59676-320) , analysis(59676-302, 59676-303, 59676-304, 59676-310, 59676-340, 59676-312, 59676-320) Establishment Name Address ID/FEI Business Operations Patheon Manufacturing Services LLC 079415560 analysis(59676-302, 59676-303, 59676-304, 59676-310, 59676-340, 59676-312, 59676-320) , manufacture(59676-302, 59676-303, 59676-304, 59676-310, 59676-340, 59676-312, 59676-320) Establishment Name Address ID/FEI Business Operations Amgen, Inc. 039976196 manufacture(59676-302, 59676-303, 59676-304, 59676-310, 59676-340, 59676-312, 59676-320) , analysis(59676-302, 59676-303, 59676-304, 59676-310, 59676-340, 59676-312, 59676-320)